Alosetron (Lotronex) wurde im Februar 2000 von der FDA zur Vermarktung zugelassen, aber im November 2000 wegen schwerwiegender, lebensbedrohlicher gastrointestinaler Nebenwirkungen vom Markt genommen. Im Juni 2002 wurde es von der FDA erneut für die Vermarktung zugelassen, jedoch in eingeschränkter Weise als Teil eines von Pharmaunternehmen gesponserten Programms zum Management der mit der Behandlung verbundenen Risiken. Die Anwendung von Alosetron ist nur bei Frauen mit schwerem Reizdarmsyndrom (IBS) mit vorherrschendem Durchfall erlaubt, die auf eine herkömmliche Behandlung von IBS nicht angesprochen haben.
Die ursprüngliche offizielle FDA-Erklärung, die herausgegeben wurde, als Alosetron ursprünglich zurückgezogen wurde, ist unten aufgeführt.
-- Medizinischer Redakteur, MedicineNet.com
28. November 2000 – GLAXO WELLCOME BESCHLIESST, LOTRONEX VOM MARKT ZU ZIEHEN Glaxo Wellcome aus Research Triangle Park, NC, hat die FDA darüber informiert, dass es Lotronex (Alosetron-Hydrochlorid)-Tabletten freiwillig vom Markt nehmen wird. Lotronex ist ein verschreibungspflichtiges Medikament, das zur Behandlung des Reizdarmsyndroms (IBS) bei Frauen zugelassen ist. Die FDA rät Patienten, die Lotronex einnehmen, sich an ihre Gesundheitsdienstleister zu wenden, um Behandlungsalternativen zu besprechen.
Die Aktion des Unternehmens folgt auf ein Treffen, das heute früher mit der Food and Drug Administration (FDA) stattfand, bei dem die Behörde mit Glaxo Wellcome Risikomanagementoptionen diskutierte, darunter die Beschränkung des Vertriebs des Medikaments oder die Rücknahme der Vermarktung.
Die heutigen Maßnahmen folgen FDA-Analysen der Post-Marketing-Berichte über schwerwiegende unerwünschte Ereignisse, darunter 5 Todesberichte bei Patienten, die Lotronex einnahmen.
Insbesondere war die FDA besorgt über gemeldete Fälle von Darmschäden, die auf eine verminderte Durchblutung des Darms (ischämische Kolitis) und einen stark verstopften oder gerissenen Darm (Komplikationen einer schweren Verstopfung) zurückzuführen sind.
Bis zum 10. November 2000 hatte die FDA insgesamt 70 Fälle schwerwiegender unerwünschter Ereignisse nach der Markteinführung erhalten und überprüft, darunter 49 Fälle von ischämischer Kolitis und 21 Fälle von schwerer Obstipation. Von den 70 Fällen führten 34 zu einem Krankenhausaufenthalt ohne Operation, 10 zu chirurgischen Eingriffen und drei zum Tod. Die FDA hat zwei weitere Todesfälle erhalten, die die Behörde nicht als Fälle von ischämischer Kolitis oder schweren Komplikationen durch Verstopfung klassifiziert hat.
Die FDA hat das Medikament seit der Zulassung am 9. Februar 2000 genau überwacht. Vor der Zulassung wurden vier Fälle von ischämischer Kolitis in klinischen Studien beobachtet und auf einer Sitzung des Gastrointestinal Drugs Advisory Committee der FDA im November 1999 diskutiert. Diese Fälle waren vorübergehend, leichter bis mäßiger Natur und reversibel nach Absetzen des Medikaments.
Zwischen der Zulassung und dem 1. Juni 2000 erhielt die FDA sieben Post-Marketing-Berichte über schwerwiegende Komplikationen der Verstopfung. Dies führte zur Hospitalisierung von sechs Patienten, von denen drei operiert werden mussten. Im gleichen Zeitraum erhielt die FDA acht Post-Marketing-Berichte über ischämische Kolitis. Dies führte zu vier Krankenhausaufenthalten, vier endoskopischen Eingriffen und keinen Operationen.
Am 27. Juni 2000 berief die FDA eine Sitzung des öffentlichen Beratungsausschusses ein, bei der Optionen für das Risikomanagement als Reaktion auf die Berichte über schwerwiegende unerwünschte Ereignisse erörtert wurden. Bis zu diesem Datum wurden keine Todesfälle gemeldet. Die Mitglieder des beratenden Ausschusses waren sich einig, dass sowohl Ärzte als auch Patienten über die potenziell schwerwiegenden unerwünschten Ereignisse im Zusammenhang mit Lotronex informiert werden müssen.
Nach dem Treffen aktualisierte die FDA die Kennzeichnung für medizinisches Fachpersonal für Lotronex und forderte den Sponsor des Medikaments, Glaxo Wellcome, auf, einen Medikationsleitfaden zu verteilen, der die Patienten direkt vor den mit dem Medikament verbundenen Risiken warnt. Darüber hinaus hat Glaxo Wellcome auf Ersuchen der FDA die Briefe „Sehr geehrter Angehöriger der Gesundheitsberufe“ und „Sehr geehrter Apotheker“ herausgegeben, um diese Gruppen über die wichtigen neuen Informationen zu informieren.
Die FDA erhielt weiterhin Berichte über schwerwiegende unerwünschte Ereignisse über ischämische Kolitis und Verstopfungskomplikationen im Zusammenhang mit Lotronex. Darüber hinaus erhielt die FDA Berichte über Todesfälle und schwerwiegendere Komplikationen der ischämischen Kolitis, die eine Bluttransfusion oder einen chirurgischen Eingriff erforderten.
Nach Abschluss ihrer jüngsten Analysen der 70 Fälle umfasste die Ansicht der FDA zu den Optionen die Marktrücknahme oder ein eingeschränktes Arzneimittelvertriebsprogramm. Das eingeschränkte Arzneimittelverteilungsprogramm würde Folgendes bieten:(1) sichere Anwendung von Lotronex bei entsprechend informierten Patienten, (2) fortgesetzten Zugang zu Lotronex für schwer geschwächte IBS-Patienten unter engmaschig überwachten Bedingungen und (3) fortgesetzte klinische Forschung zu Nutzen, Risiken, und sichere und angemessene Anwendung von Lotronex. Die FDA erkannte, dass die anderen verfügbaren Behandlungen für IBS möglicherweise keine ausreichende Linderung einer Erkrankung bieten, die für einige Patienten schwerwiegende Beeinträchtigungen darstellen kann.
Zum Abschluss des heutigen Treffens teilte Glaxo Wellcome der FDA mit, dass es Lotronex freiwillig vom Markt nehmen werde.
Weitere Informationen zu diesem Thema finden Sie auf der Lotronex-Informationswebseite, die vom FDA Center for Drug Evaluation and Research erstellt wurde. Die URL lautet www.fda.gov/cder/drug/infopage/lotronex/lotronex.htm.
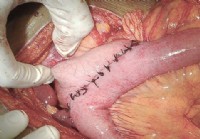 Was ist ein Gastrojejunostomie-Verfahren?
Was ist ein Gastrojejunostomie-Verfahren? Bild zeigt etwa 10 Zoll lange Nähte an inneren Organen, der Anastomose (chirurgisch geschaffene Öffnung) vom Magen bis zum Magen mittleren Dünndarm. Die Gas
Was ist ein Gastrojejunostomie-Verfahren?
Was ist ein Gastrojejunostomie-Verfahren? Bild zeigt etwa 10 Zoll lange Nähte an inneren Organen, der Anastomose (chirurgisch geschaffene Öffnung) vom Magen bis zum Magen mittleren Dünndarm. Die Gas
 Die Gehirn-Gut-Verbindung bei IBS
Eine Dysfunktion in der Verbindung zwischen Gehirn und Darm kann ein Faktor sein, der zum Reizdarmsyndrom (IBS) beiträgt. IBS ist alles andere als einfach, und Forscher blicken über den Darm hinaus un
Die Gehirn-Gut-Verbindung bei IBS
Eine Dysfunktion in der Verbindung zwischen Gehirn und Darm kann ein Faktor sein, der zum Reizdarmsyndrom (IBS) beiträgt. IBS ist alles andere als einfach, und Forscher blicken über den Darm hinaus un
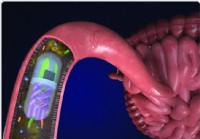 Wissenschaftler entwickeln eine 3D-gedruckte Pille, die Bakterien aus dem Darm untersucht
Das Darmmikrobiom besteht aus Billionen lebender Mikroben, und mehr als tausend Bakterienarten. Es ist bekannt, dass der Darm eine wichtige Rolle für die Gesundheit des Körpers spielt. Ein Team von Un
Wissenschaftler entwickeln eine 3D-gedruckte Pille, die Bakterien aus dem Darm untersucht
Das Darmmikrobiom besteht aus Billionen lebender Mikroben, und mehr als tausend Bakterienarten. Es ist bekannt, dass der Darm eine wichtige Rolle für die Gesundheit des Körpers spielt. Ein Team von Un