der Geschichte der Menschheit zu entdecken von Magen Bakterien
Zusammenfassung
Aktuelle Analysen menschlicher Krankheitserreger haben ergeben, dass ihre Entwicklungsgeschichte deckungsgleich sind mit dem angenommenen Muster der alten und modernen menschlichen Bevölkerung Migrationen. Die phylogenetische Bäume von Stämmen des Bakteriums Helicobacter pylori
und dem Polyoma JC-Virus aus geographisch unterschiedlichen Gruppen von Menschen genommen korrelieren eng mit den Beziehungen der Populationen, in denen sie gefunden werden.
Charles Darwin erkannte, dass die Verteilung und Form Parasiten war evolutionär signifikant. Er wies darauf hin, zum Beispiel, dass "... die Pediculinen [Läuse] in verschiedenen Ländern von den verschiedenen Menschenrassen gesammelt ... unterscheiden sich nicht nur in der Farbe, aber in der Struktur ihrer Klauen und Gliedmaßen. In jedem Fall in die vielen erhaltenen Proben wurden die Unterschiede konstant "waren [1]. In jüngster Zeit haben mehrere Forschergruppen [2-7] interessante Korrelationen zwischen den evolutionären Beziehungen zwischen verschiedenen bakteriellen und Virusstämme durch den Menschen und das Muster von Migrationen des modernen Menschen auf der ganzen Welt.
Ein besonders interessanter Fall gehostet gefunden ist, dass der Helicobacter pylori
, ein Gram-negative Bakterium im Zusammenhang mit Gastritis, Magengeschwüren und Magenkrebs, die Hälfte aller Menschen infizieren kann bis [8]. Die Entdeckung, dass eine bakterielle Infektion, was wurden als chronische Krankheiten [8] war ein eindrucksvolles Beispiel für die Tatsache führen könnten, dass Infektionskrankheiten noch nicht erobert worden. Die anhalt Acquired Immune Deficiency Syndrome (AIDS) Epidemie, Ausbrüche von Ebola in Zentralafrika, und die aktuelle Verbreitung von West-Nil-Virus in den Vereinigten Staaten und des schweren akuten respiratorischen Syndroms (SARS) aus Asien belegen die Allgegenwart und gesundheitlichen Folgen von Infektionserreger auch im Zeitalter der Impfung und antimikrobielle und antivirale Therapien. Viele Infektionskrankheiten sind gedacht, um gleichzeitig mit der Entwicklung der Landwirtschaft und der Aufstieg des städtischen Lebens entstanden. Wenn stattdessen sind viele Krankheitserreger "Beziehungen mit den Menschen viel älter, würde es nicht überraschen, zu tiefer evolutionären Verbindungen zwischen Menschen und ihrer mikrobiellen und viralen Eindringlingen finden.
Die Evolutionsgeschichte von H. pylori Du ein Beispiel liefern kann der Koevolution eines Bakteriums und seine einzige bekannte Wirt. Die H. pylori
Genom ist relativ klein bei 1,67 Megabasen, mit einer minimalen Komplement von metabolischen Gene [9]. Variation zwischen H. pylori
Isolate von verschiedenen Personen oder auch von einer Person ist groß, was zu einer einzigartigen Fingerabdrücke für fast jedes Isolat bisher eingegeben haben. Die kodierenden Gene sind nicht sehr vielfältig, aber: Die meisten der Variation tritt in der dritten Grundposition innerhalb Codons oder durch Inversionen oder translo-Kationen, die codierten Aminosäuresequenzen relativ ähnlich zu verlassen [2]. Diese Aminosäuresequenz Erhaltung ist für die Impfstoffforschung Glück, als ein Impfstoff ist wahrscheinlich auf viele Stämme wirksam zu sein. Interessanter ist die Tatsache, dass H. pylori
eine extrem hohe Rate der Rekombination hat, höher in der Tat als die jeder anderen bisher gekennzeichnet Organismus [3, 10].
Normalerweise ist eine solche hohe Rate der Rekombination würde machen eines Organismus Evolutionsgeschichte sehr schwierig, da die Informationen über die Herkunft der einzelnen Mutation folgern verloren gehen würde, wie sie während einer Bevölkerung verbreitet. Aber in Verbindung mit der Art der Übertragung von H.-pylori-
, diese extrem hohe Rate der Rekombination kann in der Tat evolutionär Schlüsse zu erleichtern. Mehrere Studien legen nahe, dass H. pylori
in der Regel innerhalb von Familien übertragen wird, in der Regel von der Mutter auf das Kind [11, 12]. Somit ist die Übertragung von H. pylori
in mancher Hinsicht ahmt die mater übertragen mitochondrialen DNA [13]. Weil Mitochondrien-DNA ausschließlich von einem Elternteil übertragen wird (der Mutter) und rekombinieren nicht, hat es sich als ideal, genetisches System zur Ableitung menschlichen Evolutionsgeschichte (siehe unten) [14, 15] zu sein. Wenn H. pylori
ist in der Tat überwiegend maternal übertragen werden neue Stämme im Allgemeinen nicht eine Person im Laufe ihres Lebens infizieren; würde dies bedeuten, zusammen mit der hohen Rate der Rekombination, dass die Mutationen, die innerhalb der Population von Bakterien in einer Person Magen ansammeln relativ homogen sein. Dies sollte in einem Schwarm von Stämmen führen, die zueinander sehr eng verwandt sind, sind viele der Mutationen enthalten, die in einzelnen Bakterien aufgetreten sind. Schwärme in verschiedenen Menschen gefunden wird somit mehr voneinander verschieden, als wenn es weniger Rekombination.
Die meisten Infektionskrankheiten schnell in der ganzen Welt verbreitet und Stämme aus verschiedenen Regionen sind relativ ähnlich, aber Erstbemusterung von H. pylori
von Menschen aus verschiedenen Regionen der Welt zeigte, ziemlich starke geografische Aufteilung in europäischen und asiatischen H. pylori
Typen [2-4]. Vor kurzem Falush und Kollegen [5] haben diese Aufteilung näher untersucht. Nach der Sequenzierung acht Gene - insgesamt 3850 Nukleotiden - in 370 von 27 menschlichen Populationen abgeleitete Stämme, fanden sie 1418 polymorphe Nukleotid-Positionen. Sie wendeten dann ein neues analytisches Werkzeug, STRUKTUR [16], das entwickelt wurde, menschliche genetische Struktur von multilocus Genotyp Daten abzuleiten. Das Programm nutzt Bayes-Methoden Untergruppen mit charakteristischen Allelfrequenzen zu identifizieren und Cluster der Untergruppen, auch in Gegenwart von Rekombination [16]. Wenn diese Technik auf die H. pylori
Sequenzen angelegt wurde, wurden vier Hauptcluster gefunden - zwei aus Afrika und jeweils einer aus Europa und Asien (Abbildung 1a) [5]. Abbildung 1 Die Beziehungen zwischen menschlichen Populationen, wie von H. pylori berechnet
Mägen und von mitochondrialer DNA-Daten gefunden. (A) Die Beziehungen zwischen den modernen Subpopulationen von H. pylori
[5]. Jede Subpopulation wird durch einen Kreis mit einem Durchmesser proportional zur genetischen Vielfalt darin vertreten. Die Zentren der Kreise sind durch einen phylogenetischen Baum, die die Beziehung zwischen den vier Subpopulationen verbunden. Bakterien in jeder Subpopulation werden vor allem in Menschen gefunden, die aus den Regionen stammen gezeigt. (B) Ein Bevölkerungsebene phylogenetischen Baum der geographischen Subpopulationen
H. pylori in (a). (C) ein Median-Verbindungsnetzwerk von menschlichen Populationen von mitochondrialer DNA abgeleitet [14]. Ein solches Netzwerk zeigt alternative potenzielle evolutionären Beziehungen zwischen den Clustern. Jeder Kreis steht für eine Gruppe von mitochondrialen Typen mit einem Durchmesser proportional zur Frequenz dieses Typs innerhalb der Subpopulationen. Alle nicht-afrikanischen Bevölkerung sind von einer afrikanischen Abstammung abgeleitet; das Netz der Beziehungen innerhalb dieser Linie ist (oben) vergrößert. (A, b) Übernommen aus [5]; (C) adaptiert von [14]
Jeder Cluster von Falush und Kollegen festgestellt, [5] in Untergruppen aufgeteilt werden könnten. zum Beispiel könnte der "Afrika 1 'Cluster weiter in West-und South African Subclustern unterteilt werden, und der Ostasien-Cluster in ostasiatischen, Amerind und Maori Subclustern aufgeteilt werden könnte. Die geografische Aufteilung innerhalb der 200 europäischen Stämme war besonders kompliziert, vermutlich deshalb, weil zahlreiche Gruppen hin und her in ganz Europa in den vergangenen Jahrtausenden gefegt. Europäische Stämme auch in Nord- und Südamerika, Australien gelegentlich erschienen und unter Südafrikaner, vermutlich koloniale Eroberung widerspiegelt., Die Verwandtschaftsverhältnisse dieser Cluster (Abbildung 1b) und ihrer Untergliederungen [5] zeigen ein ähnliches Muster wie bei diesem, erhalten unter Verwendung der mitochondrialen DNA Variante (1c) [14, 15]. Der moderne Mensch Genpool, wie aus der mitochondrialen DNA entnommen und durch Y-Chromosom-Studien bestätigt, wird angenommen, dass etwa 150.000-200.000 Jahren einen afrikanischen Ursprung gehabt zu haben [14, 15, 17, 18]. Die ursprüngliche menschliche Bevölkerung dann verbreitet und in ganz Afrika für fast 100.000 Jahren diversifiziert, bevor sie in Westasien und Europa zu erweitern und in Süd- und Ost-Asien vor ca. 50.000-60.000 Jahren in diesen Regionen die bestehenden archaischen Populationen von Menschen zu ersetzen. Nachfolgende Migrationen verbreitet in Australien von vor 40.000 Jahren, dann zu den Pazifischen Inseln und später in Nordamerika, etwa vor 15.000 Jahren (Abbildung 2) [14, 15, 17, 18]. Die bemerkenswerte Ähnlichkeit zwischen dieser Sicht der menschlichen Geschichte und die Ergebnisse aus Untersuchungen von H. pylori
haben dazu geführt, Falush und Kollegen [5] sowie andere [2] zu dem Schluss, dass H. pylori
Evolution den Weg verfolgt hat, der modernen menschlichen Expansion und Migration. Diese Arbeit stellt somit eine andere Art von Daten für die Analyse der menschlichen Evolution und Migration, unabhängig von mitochondrialer DNA und Y-Chromosomen, die in weiteren Untersuchungen wertvoll sein wird. Leider jedoch stammt aus H. pylori
die Divergenz der Schätzung ist besonders schwierig, die extrem hohe Rate der Rekombination wegen [10]. Zusätzliche Probenahme- und Analysetechniken können die Migrations Hypothese weiter zu testen, erforderlich. Figur 2 Eine Karte des Musters der Expansion und Migration des modernen Menschen in der ganzen Welt, abgeleitet aus Untersuchungen der mitochondrialen DNA und Y Chromosomen [14, 15, 17, 18]. Die Zahlen geben die ungefähre Zeit (in Jahren vor der Gegenwart), wenn der moderne Mensch zuerst in der angegebenen Region erschienen.
Andere Krankheitserreger auch Entwicklungsgeschichte zu folgen ähnlich wie ihre Wirte vorgeschlagen worden. Eine der interessantesten, wenn auch mit einem nicht-menschlichen Wirt ist, dass der Blattläuse, ein Bakterium in ihnen gefunden, und zwei mit dem Bakterium assoziierten Plasmiden. Funk et al.
[19] festgestellt, dass die abgeleiteten intraspecifrc phylogenies dieser vier Genome vollständig deckungsgleich waren. Rückkehr zum menschlichen Krankheitserreger, kann das menschliche Polyomavirus JC-Virus (JCV) in Genotypen unterteilt werden, die zu den großen kontinentalen Landmassen entsprechen [6]. Wie H. pylori
, JCV - die progressive multifokale Leukoenzephalopathie (Verlust der Myelinisierung im zentralen Nervensystem) verursachen kann - ist sehr weit verbreitet unter den Menschen als Folge der familiären Übertragung. Insgesamt 12 bekannten Subtypen wurden definiert, mit europäischen, afrikanischen und asiatischen Verteilungen [7]. Obwohl eine direkte Schlüsse eines afrikanischen Ursprungs für JCV problematisch sind, weil es keine geeigneten outgroup ist, mit dem der phylogenetischen Baum zu verankern, wenn ein afrikanischer Herkunft angenommen wird, kann eine vernünftige evolutionäre Geschichte vermutet werden (Abbildung 3) [7]. Wie bei H. pylori
stammt Molekular Divergenz Folgern ist derzeit problematisch für JCV. Weitere Untersuchungen in die Evolutionsgeschichte dieser menschlichen Krankheitserreger ist daher notwendig. Abbildung 3 Relationships menschlicher Polyoma JC-Virus (JCV) beim Menschen aus verschiedenen Teilen der Welt gefunden Subtypen [7]. Die Buchstaben beziehen sich auf einzelne Subtypen. (A) Die Hypothese Muster der Verbreitung von JCV-Subtypen durch die Welt (mit Ausnahme von Nord- und Südamerika); (B) eine abgeleitete Phylogenie von JCV-Subtypen, einen afrikanischen Ursprung für das Virus übernehmen. Übernommen aus [7].
Die Muster der Evolution der menschlichen Krankheitserreger Aufklärung letztlich kann zusätzliche Beweise nicht nur über ihre Geschichte, sondern auch über die menschliche Evolution und Geschichte. Dies wird vor allem für Krankheitserreger wie H. pylori
, die eine überwiegend Mutter-Kind-Modus der Übertragung haben, mitochondriale DNA Evolution nachahmt. H. pylori
's ursächliche Rolle bei mehreren chronischen Magen Bedingungen ist nur eine von vielen aktuellen Beispiele für die bekannten oder vermuteten Rolle eines Erregers zu chronischen Krankheiten führen. Die Bakterien werden im Verdacht bei der Entwicklung von Arteriosklerose, Schlaganfall und Morbus Crohn beteiligt zu sein, während Viren bekannt sind, AIDS und die verschiedenen Formen der chronischen Hepatitis führen. Gebärmutterhalskrebs, hepatozellulärem Karzinom, Burkitt-Lymphom, Kaposi-Sarkom, und vielleicht mellitus Diabetes sind ebenfalls entweder bekannt oder vermutet viralen Ursprungs sein. Wenn einige der allgegenwärtigen chronischen Krankheiten beweisen bakteriellen oder viralen Ursprungs, weltweite Erhebungen dieser Erreger in menschlichen Populationen zu sein, sollte sofort durchgeführt werden, so dass das Wissen über die Evolution und Diversität dieser Erreger können in die Forschungsprogramme integriert werden entwickelt, um verbessern die Bedingungen, die sie verursachen.
 Dysbiose in der Darmmikrobiota kann bei COVID-19-Patienten schwere Sekundärinfektionen verursachen
Dysbiose in der Darmmikrobiota kann bei COVID-19-Patienten schwere Sekundärinfektionen verursachen
 Die westliche Ernährung kann das Risiko einer „tödlichen Sepsis“ erhöhen.
Die westliche Ernährung kann das Risiko einer „tödlichen Sepsis“ erhöhen.
 Neugeborenes Mausmodell liefert Hinweise auf die Ursache einer verheerenden Darmerkrankung bei anämischen Frühchen
Neugeborenes Mausmodell liefert Hinweise auf die Ursache einer verheerenden Darmerkrankung bei anämischen Frühchen
 Wissenschaftler entwickeln einen Ansatz zur Impfung gegen Darmentzündungen
Wissenschaftler entwickeln einen Ansatz zur Impfung gegen Darmentzündungen
 Mundwasser beeinflusst die Wirkung von Bewegung
Mundwasser beeinflusst die Wirkung von Bewegung
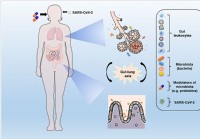 Die Modulation der Mikrobiota und die Wiederherstellung der Eubiose könnten helfen, COVID-19-Komplikationen einzudämmen
Die Modulation der Mikrobiota und die Wiederherstellung der Eubiose könnten helfen, COVID-19-Komplikationen einzudämmen
 Wissenschaftler beweist Rolle des Mikrobioms bei Fettleibigkeit
Neue Forschungsergebnisse von Dr. Christoph Thaiss in seinem preisgekrönten Essay zum „ Wissenschaft und SciLifeLab Prize for Young Scientists“ beschreibt, dass mehrere bakterielle Stoffwechselprodu
Wissenschaftler beweist Rolle des Mikrobioms bei Fettleibigkeit
Neue Forschungsergebnisse von Dr. Christoph Thaiss in seinem preisgekrönten Essay zum „ Wissenschaft und SciLifeLab Prize for Young Scientists“ beschreibt, dass mehrere bakterielle Stoffwechselprodu
 Die mediterrane Ernährung fördert ein gesundes Altern mit einem gesünderen Darmmikrobiom
Eine neue Studie, die online in der Zeitschrift veröffentlicht wurde Darm berichtet im Februar 2020 über die markanten gesundheitsfördernden Effekte einer Umstellung auf eine mediterrane Ernährung f
Die mediterrane Ernährung fördert ein gesundes Altern mit einem gesünderen Darmmikrobiom
Eine neue Studie, die online in der Zeitschrift veröffentlicht wurde Darm berichtet im Februar 2020 über die markanten gesundheitsfördernden Effekte einer Umstellung auf eine mediterrane Ernährung f
 Wenn Sie über 50 sind,
Es ist Zeit für eine Koloskopie Die Leute schieben bestimmte Dinge auf – die Garage ausräumen, Wohnzimmer neu streichen, Fenster putzen ... und eine Darmspiegelung machen lassen. Aber eine Koloskopie
Wenn Sie über 50 sind,
Es ist Zeit für eine Koloskopie Die Leute schieben bestimmte Dinge auf – die Garage ausräumen, Wohnzimmer neu streichen, Fenster putzen ... und eine Darmspiegelung machen lassen. Aber eine Koloskopie