Résumé
Microarray des données d'expression génique de 54 cancer gastrique associé et les tissus gastriques adjacents noncancerous ont été analysés dans le but d'établir des signatures génétiques pour grades de cancer (bien-être, moderately-, poorly- ou non différencié) et les étapes (I, II, III et IV), qui ont été déterminées par les pathologistes. Notre analyse statistique a conduit à l'identification d'un certain nombre de combinaisons de gènes dont l'expression patterns servir ainsi que les signatures de différentes qualités et différents stades du cancer gastrique. Une signature de 19 gènes a été constaté que le pouvoir discerner entre les cancers gastriques haute et basse qualité en général, avec une précision globale de classification à 79,6%. Un groupe de 198 gènes élargi permet la stratification des cancers en quatre catégories et le contrôle, ce qui donne lieu à un accord de classement général de 74,2% entre chaque grade désigné par les pathologistes et notre prédiction. Deux signatures pour la stadification du cancer, composé de 10 gènes et 9 gènes, respectivement, fournissent des exactitudes de haute classification à 90,0% et 84,0%, entre, cancer avancé stade précoce et de contrôle. Fonctionnel et voie analyses sur ces gènes de signature révèlent la pertinence significative des signatures provenant de grades de cancer et de progression. Au meilleur de notre connaissance, ce qui représente la première étude sur l'identification de gènes dont l'expression patterns peuvent servir de marqueurs pour les grades et stades cancer
Citation:. Cui J, Li F, Wang G, Fang X, Puett JD, Xu Y (2011) Signatures Gene expression peut distinguer les qualités et les stades du cancer gastrique. PLoS ONE 6 (3): e17819. doi: 10.1371 /journal.pone.0017819
Editeur: Amanda Toland, Medical Center Ohio State University, États-Unis d'Amérique
Reçu: 24 Novembre 2010; Accepté 9 Février 2011; Publié le 18 Mars, 2011
Droit d'auteur: © 2011 Cui et al. Ceci est un article en accès libre distribué sous les termes de la licence Creative Commons Attribution, qui permet une utilisation sans restriction, la distribution et la reproduction sur tout support, à condition que l'auteur et la source originelle sont crédités
Financement:. Cette étude a été soutenu en partie par la national science Foundation (DEB-0830024, DBI-0542119), les national Institutes of Health (1R01GM075331), un «Distinguished Scholar" subvention du Cancer Coalition Géorgie, et un fonds d'amorçage conjointement par Venture Fund du président et le Bureau du vice-président pour la recherche de l'Université de Géorgie. Les bailleurs de fonds ont joué aucun rôle dans la conception de l'étude, la collecte et l'analyse des données, la décision de publier, ou de la préparation du manuscrit
Intérêts concurrents:.. Les auteurs ont déclaré aucun conflit d'intérêts existent
Introduction
classement du cancer est une mesure de la malignité et l'agressivité d'un cancer. Un système de classement populaire utilise quatre niveaux de malignité (G1-G4), ce qui reflète le niveau combiné de cellules apparence d'anomalie, l'écart du taux de croissance des cellules normales et le degré d'invasivité et la diffusion. Ces mesures pathologiques ont été trouvés pour être en concordance générale avec le niveau de la différenciation cellulaire (Commission mixte américaine sur le cancer) [1]. Ainsi {G1, G2, G3, G4} sont aussi appelé bien, moderately-, poorly- et non différenciés, respectivement. A partir de maintenant, il n'y a pas eu un système de classement universel pour tous les cancers. Au lieu de cela, les systèmes de notation différents ont été proposés pour différents cancers. Par exemple, le système de Gleason [2] est probablement le plus connu pour le classement des cellules d'adénocarcinome dans le cancer de la prostate alors que le système Bloom-Richardson [3] est utilisé pour le cancer du sein, et le système Fuhrman [4] est utilisé pour le cancer du rein .
le cancer gastrique, la deuxième principale cause de décès liés au cancer dans le monde entier, est particulièrement répandue dans les pays asiatiques, dont la Chine, la Corée et le Japon [5]. Aux États-Unis, cette maladie asymptomatique avait ~21,500 nouveaux cas en 2008 avec 10.800 décès [6]. Contrairement à d'autres cancers, le cancer gastrique n'a encore un système de classement généralement acceptée. Le classement a été fait principalement sur la base des lignes directrices générales plutôt cancer classement des organisations comme la Commission mixte américaine sur le cancer. Il y a quelques systèmes de classification des cancers gastriques en sous-types histologiques, y compris ceux de la Lauren [7], l'Organisation mondiale de la santé (OMS) [8] et Goseki, et al. [9], [10], qui définissent des sous-types en fonction des caractéristiques structurelles du cancer, les apparitions histopathologiques des cellules, et le niveau de mucus, respectivement. Cependant, il est largement controversé quant à savoir si l'un de ces systèmes est vraiment pertinent pour le degré de malignité et de survie, ainsi ne pas avoir été largement utilisé pour le classement du cancer gastrique [11]. Le manque d'un système de classement bien établi pour le cancer gastrique reste un obstacle majeur entravant les progrès dans ce domaine.
Nous présentons une étude computationnelle ici, visant à identifier un ensemble de gènes dont l'expression patterns peut bien distinguer parmi les cancers gastriques de différentes qualités, comme Oncotype DX, un panneau de 21 gènes pour identifier le cancer du sein à faible risque [12]. Ces gènes, dont l'expression patterns distinguer les cancers gastriques de différents grades, fournissent des informations utiles à l'élaboration d'un système de classement basé expression génique pour le cancer gastrique. En outre, nous présentons également nos conclusions sur les profils d'expression des gènes communs aux cancers à différents stades de développement, ce qui pourrait servir les signatures moléculaires pour la stadification du cancer gastrique.
Résultats. L'identification des gènes avec des changements d'expression en corrélation avec
17.800 gènes humains de cancer grades ont été profilées dans cette étude, en utilisant Affymatrix Exon Arrays. Sur les échantillons de cancer 54, 8 sont bien différenciés (DEO), 9 modérément différenciés (MD), 35 peu différencié (PD) et 2 indifférencié (UD). Un total de 452 gènes ont été trouvés être exprimé de manière différentielle tel que déterminé selon les critères suivants: les niveaux d'expression dans le cancer et l'exposition des tissus de contrôle correspondant de changement au moins 2 fois, et la signification statistique, P Nous avons ensuite vérifié si certains gènes peuvent avoir leurs changements d'expression sont en corrélation avec les classes de cancer. Pour ce faire, nous avons calculé le coefficient de corrélation de Spearman (CC) entre l'expression moyenne de chaque gène dans tous les échantillons de chaque grade et les quatre catégories de cancer. Il a été constaté que les changements d'expression de 99 gènes corrèlent parfaitement avec les grades WD-MD-PD-UD (| CC Nous avons constaté que les gènes avec leurs changements d'expression en corrélation avec les qualités de cancer sont fortement enrichis parmi les protéines sécrétées ou membranaires. (valeur P < 0,05) , qui participent à de multiples voies de signalisation telles que ErbB, FAS, récepteur NOD-like, PPAR et signalisation Wnt, ainsi que des molécules d'adhésion cellulaire (CAMs) et les jonctions serrées. Cela ne veut pas surprenant étant donné que ces voies sont essentiellement impliquées dans la croissance cellulaire et la mort cellulaire, ainsi que les métastases du cancer. De tels changements dans les modes d'expression génique de ces voies, impliquées dans la transduction du signal et de la communication extracellulaire, peuvent fournir des indices sur la progression du cancer. Nous avons examiné les 452 gènes différentiellement exprimés, visant à identifier les gènes dont l'expression peut modèles, avec une bonne précision et de la fiabilité, de distinguer les cancers gastriques de différentes qualités. L'analyse de classification (voir Méthodes) a été réalisée entre deux groupes de cancer (très et peu différenciées), puis étendue à cinq groupes, à savoir quatre grades de cancer et le contrôle. Une machine à vecteurs de support (SVM) à base régressive approche fonction d'élimination a été appliquée, en utilisant un noyau linéaire pour la classification du cancer (voir Méthodes). A la fin, un groupe de 19 gène a été identifié qui peut distinguer les très et mal cancers avec un accord global différencié à 79,2%, sur la base du pli de changement d'expression dans le cancer par rapport La signature de 19 gènes se compose de ADIPOQ, COL6A3, TNS1, SCN7A, dES, VIL1, COL3A1, C2orf40, SMYD1, ACTG2, MEIS1, C7, GPR174, SHCBP1, DUSP1, DNAJB5, HIATL1, IL17RB et FAT. Un examen attentif de l'annotation fonctionnelle de ces gènes a révélé que leurs produits protéiques sont impliqués dans la croissance et la différenciation cellulaire (IL17RB, SMYD1, SHCBP1), la motilité cellulaire (ACTG2), l'angiogenèse et le remodelage des tissus (ADIPOQ), carcinogenèse (ECRG4), matrice la synthèse des protéines (COL3A1, COL6A3), et d'autres comme G récepteur couplé à la protéine 174 (GPR174), cytosquelette de bordure en brosse (VIL1), complexe d'attaque membranaire (C7), et le canal de sodium (SCn7A). 17 sur des 19 gènes, plus un 181 gènes supplémentaires, forment un groupe de 198 gènes dont le motif d'expression peut distinguer les quatre catégories de cancer et le contrôle. Leurs fonctions couvrent la division cellulaire, la réponse immunitaire, la transduction du signal et de régulation de la transcription, en plus des catégories mentionnées ci-dessus. Dans l'ensemble, 39 des 99 gènes de qualité corrélés font partie de cette signature de 198 gènes, y compris CLDN1, MUC13, VIL1, HIATL1, CDCA7, HIST1H2BM et FAT (voir la liste complète dans le tableau S3). outre ce fourre-tout la signature pour la classification à cinq voies, nous avons également identifié et analysé les signatures de gènes spécifiques de qualité pour chaque catégorie de cancer. Par exemple, LAPTM4B est un tel représentant. Ce gène donne une grande précision de classification pour caner et de contrôle des échantillons dans le groupe WD avec l'AUC (aire sous la courbe) = 0,97 (figure 3). Utilisation de 7,04 comme seuil d'expression, ce gène peut bien distinguer le cancer à partir des échantillons de contrôle dans le groupe WD avec une sensibilité = 87,5% et une spécificité = 100%. Ce résultat est pas surprenant puisque l'on sait que LAPTM4B est essentielle pour la croissance et la survie cellulaire, et sa régulation positive a été trouvée être en corrélation avec le degré de différenciation d'un carcinome hépatocellulaire [15]. Au total, 40 de ces gènes de signature se trouvent spécifiquement pour le groupe WD; 18, 20 et 255 gènes sont spécifiques au groupe MD, PD et UD, respectivement (voir les détails dans le tableau S4). Nous avons également identifié discriminateurs un seul gène pour chaque groupe de qualité contre le reste des échantillons, y compris le contrôle, tel que résumé dans le tableau 1. par exemple, les signatures pour le groupe PD comprennent les gènes régulés à la hausse, MYO1B C. Identification des signatures de gènes pour le stade Utilisation des analyses similaires à celles de ce qui précède, nous avons identifié les signatures de gènes pour stade précoce (stade I + II) et le cancer de stade avancé (stade III + IV). Le tableau 2 met en évidence des marqueurs les plus discriminantes monogéniques, avec la précision de classification allant de 75,0% à 81,4%. signatures multi-gènes ont également été vérifiées pour la stadification du cancer. Par exemple, deux signatures se sont révélés être particulièrement efficaces pour la stadification du cancer, à savoir un groupe de 10 gènes (CPS1 + DEFA5 + DES + DMN + GFRA3 + MUC17 + OR9G1 + REEP3 + TMED6 + TTN) et un groupe 9-gène (DPT + EIF1AX + FAM26D + IFITM2 + LOC401498 + OR2AE1 + PRRG1 + REEP3 + RTKN2) , ce qui permet de distinguer le début et les cancers gastriques avancés à partir du reste des échantillons (y compris les échantillons témoins) avec les accords de 90,0% et 84,0%, respectivement. La précision globale de classification sur les trois groupes, au début, avancée et de contrôle, est de 71,4%. Une analyse fonctionnelle sur ces gènes de signature a révélé quelque chose d'intéressant. Par exemple, parmi les produits de protéines de signature gènes précoce de la scène, GFRA3 Quelques gènes ont été trouvés pour être à la fois le classement du cancer et signatures mise en scène, comme CPS1, dES, GFRA3, TMED6 et DPT, ce qui indique une certaine pertinence biologique entre la différenciation du cancer et de la progression. Nous avons ensuite examiné si l'expression du gène de signatures d'arrêt sont associés à des stades pathologiques. Parmi eux, ceux qui sont très corrélés à différents stades pathologiques sont LANCL3 D. Identification des gènes différentiellement exprimés indépendants de grades et stades cancer En plus de l'expression différentielle spécifiques à certains sous-groupes de cancer de l'estomac, nous avons également examiné si certains gènes sont exprimés différemment dans le cancer gastrique en général, indépendamment des grades et les étapes. 62 de ces gènes ont été trouvés avec l'expression différentielle cohérente par au moins les changements de 2 fois dans le cancer par rapport Seulement trois, CLDN7 Les profils d'expression de nos gènes de signature identifiés ont été vérifiés contre deux ensembles de données publiques, à savoir, le Kim dans l'ensemble, notre signature de 19 gènes pour les grades de cancer ont bien performé sur le Kim Fait intéressant, nous avons constaté qu'il existe une corrélation modérée entre l'expression du gène de notre groupes de signature identifiés et la récurrence du cancer sur la base des informations rechute péritonéale des données de Takeno [24]. Plus précisément, les quatre signatures, 19-, 198-, 10 et 9 gènes groupes, peuvent prédire la rechute péritonéale avec une précision globale de 66,0%, 87,2%, 73,0% et 55,3%, respectivement, en distinguant entre les des rechutes patients libres et péritonéale-rechute dans l'étude de Takeno [24]. analyses Discussion biopuces d'expression du gène sur le cancer gastrique ont déjà identifié les profils d'expression génique pour la prédiction de pronostic [25], [26] et le diagnostic général de cancer [27], [28] (tel que revu dans le tableau S6), mais aucune pour le sous-typage de cancer de l'estomac ou le classement. Ici, nous avons présenté une analyse sur 54 paires de cancer et les tissus de référence adjacents du même nombre de patients atteints de cancer gastrique, et les signatures moléculaires identifiées pour les grades et stades cancer. Il est connu que les différentes analyses de sélection de classification et de gènes peut conduire à différentes signatures de gènes, ce qui pose un problème sérieux au sujet de la stabilité et l'utilité des signatures de gènes sélectionnés. Pour faire face à ce problème, nous avons appliqué des recherches exhaustives pour les signatures k-géniques (k < = 4) couplée à une procédure de sélection de fonctionnalités robustes avec vote à la majorité pour k > 4, qui assure la stabilité des gènes de signature identifiés. D'autre part, en raison de la nature complexe des données du cancer d'expression génique, une croyance générale a été que les différentes techniques de classification peuvent donner lieu à des signatures différentes mais d'égale importance, car ils peuvent correspondre à différentes voies associées à différents aspects d'un cancer . En plus de ces écarts techniques, la taille limitée de l'échantillon et de l'hétérogénéité qui existe entre les sous-groupes de cancer sont notés comme d'autres facteurs importants affectant les marqueurs sélectionnés. En conclusion, nous avons démontré ici que les profils d'expression génique peuvent être utilisés que les signatures efficaces pour le classement gastrique du cancer et la mise en scène, ainsi que la prédiction pronostique. Deux types de signatures ont été proposés pour servir à des fins de diagnostic différentes, chacune représentant une certaine importance pour malignance du cancer et la progression du cancer. De telles tentatives d'utilisation de qualité et stade signatures moléculaires devraient bénéficier de manière significative le développement de la médecine personnalisée et peuvent conduire à de nouveaux marqueurs sériques. Des échantillons de tissus de Matériels et méthodes des échantillons ont été prélevés dans les cancers gastriques malignes primaires de patients non traités au cours de l'intervention chirurgicale initiale à trois hôpitaux affiliés du Collège de médecine et de cancérologie de l'Hôpital provincial de Jilin, Changchun, Chine Université de Jilin. Pour chaque échantillon de tissu de cancer, un échantillon de tissu de référence correspondant a été recueilli à partir de la région adjacente noncancerous que le chirurgien réséquée afin d'assurer une marge positive. Tous les échantillons ont été soumis à une congélation dans l'azote liquide à moins de 10 minutes après l'excision et stockées à -196 ° C jusqu'à l'extraction de l'ARN. Pour l'isolement de l'ARN, 100 um sections de chaque échantillon ont été utilisés. Tous les dossiers médicaux et les coupes de cancer ont été examinés par un pathologiste chirurgicale, et le diagnostic histologique et la classification TNM ont été faites selon organisation mondiale de la santé (OMS) critères et le système de l'Union internationale contre le cancer de la classification. Les échantillons de référence ont été soumises à une analyse histologique minutieuse afin de garantir l'absence totale de cellules cancéreuses. Le consentement éclairé écrit a été obtenu de tous les patients, qui a été approuvé par le comité d'examen institutionnel à l'Université de Géorgie, Athens, Géorgie, Etats-Unis et par la CISR chinoise superviser des sujets humains à Jilin University College of Medicine et de l'Hôpital du cancer de Jilin Provincial, Changchun , Chine. des informations détaillées des patients tels que l'âge, le sexe, le type histologique, grade différentiel, stade pathologique et de l'histoire de l'utilisation de l'alcool /tabac est listé dans le tableau S5. biopuces les échantillons d'ARN ont été analysés en utilisant le GeneChip Human Exon 1.0 ST (Affymetrix), suivant le protocole détaillé dans l'expression GeneChip Manuel Analyse technique (P /N 900223) pour l'expérience de tableau et un rapport antérieur [29]. Les microréseaux ont été numérisés à l'aide du scanner GeneChip 3000 avec GeneChip Operating Software (SMOC). Toutes les données sont MIAME conformes et les données brutes a été déposé dans la base de données GEO (ID: GSE27342). résultats de l'expression de gènes ont été résumées sur la base des intensités de sonde brutes en utilisant le multipuce robuste moyenne [30] et le package APT (http://www.affymetrix.com/partnerSupplementaryprograms/programs/developer/tools/powertools.affx), après trois étapes principales, y compris la correction de fond, la normalisation quantile et log2-transformation. Des gènes ayant une très faible expression dans les échantillons de cancer et de référence ont été enlevés; spécifiquement, un gène a été retiré si son maximale (Expr.cancer, Expr.normal) Deux stratégies différentes ont été appliquées pour évaluer l'importance des gènes, selon à quelles conditions ont été comparées et si échantillons appariés ou non appariés doivent être utilisés. Pour la comparaison des cancers contre des groupes d'échantillons de contrôle, des tests non appariés ont été menées pour vérifier si deux groupes d'expression sont différents, alors que les tests appariés ont été appliquées pour examiner la cohérence des changements d'expression à travers toutes les paires. Outre le Wilcoxon test, nous avons aussi appliqué un autre test statistique simple pour détecter des gènes avec une expression différentielle cohérente dans le cancer par rapport Pour k signatures -Gene (k < = 4), nous avons effectué une recherche exhaustive pour toutes les combinaisons k-géniques entre les gènes exprimés de manière différentielle, identifiés à partir de l'étape précédente, en utilisant une approche de classification basée sur SVM linéaire, et la précision globale a été évaluée à l'aide 5 fois la validation croisée. Pour k > 4, une approche différente à l'aide d'une recherche heuristique a été appliquée depuis la recherche exhaustive est trop de temps pour être pratique pour notre problème. Les détails sont les suivants. L'ensemble des données d'expression a été divisé au hasard en apprentissage et de test, contenant chacun la moitié des échantillons. Cela a été répété pour 500 fois pour générer 500 ensembles de données de formation /d'essai pour la classification. Un SVM linéaire a été utilisée pour la formation d'un classificateur [31], [32]. Elle construit un hyperplan qui sépare deux différentes classes de vecteurs de caractéristiques avec une marge maximale. Cette hyper-plan est construite en trouvant un vecteur w b et d'une variable qui réduisent au minimum, ce qui satisfait aux conditions suivantes: , pour (échantillons de cancer) et (échantillons normaux). Ici, est un vecteur de caractéristique est l'index de groupe, w est un vecteur normal à l'hyper-plan est la distance de l'hyperplan à l'origine et qui est la norme euclidienne de p. Après la détermination des valeurs w et b, un vecteur x donné peut être classé en utilisant; une valeur positive ou négative indique que le vecteur x appartient à la classe positive ou négative, respectivement. signatures génétiques de chaque ensemble de formation ont été sélectionnés en utilisant la procédure fonction d'élimination récursive (RFE), qui est un wrapper qui sélectionne les gènes prédictives en éliminant les gènes non-prédictives selon une fonction de gène rang généré à partir du système de classification [33]. Le critère de classement est basé sur la variation de la fonction objective lors du retrait de chaque gène. Pour améliorer l'efficacité de la formation, cette fonction objective est représentée comme une fonction de coût J Les 500 ensembles de formation /d'essai ont été répartis au hasard en 10 groupes d'échantillons. Chaque groupe d'échantillons a ensuite été utilisé pour obtenir une signature, sur la base du vote à la majorité et l'évaluation de la cohérence des gènes de rang dans les 50 formations et de test. Les 10 signatures différentes dérivées des 10 groupes ont été comparés pour évaluer le niveau de cohérence entre les gènes sélectionnés. Dans chaque groupe, des sous-ensembles de gènes ont été sélectionnés par RFE-SVM de chaque série de formation, ainsi que la performance sur les sous-ensembles a été évaluée à partir de l'ensemble de test associé. Pour obtenir un gène critère de classement uniforme pour toutes les itérations, une RFE fonction de classement à chaque étape d'itération a été dérivé d'un classificateur SVM qui a donné la meilleure précision de la classification moyenne sur les ensembles 50 de test. Deux ensembles de données de microréseaux publics ont été téléchargés à partir de la base de données GEO pour des études comparatives, le Kim
-value , d'avoir ce niveau de changement d'expression est < 0,05 (voir Matériel et méthodes, les noms de gènes sont énumérés dans le tableau S1). Parmi les 452 gènes, 97 uniquement dans UD, 62 PD, 8 MD et 16 uniquement dans WD représentent un core ensemble
des gènes exprimés de manière différentielle, qui sont toujours identifiés par l'application de différentes stratégies de classification en utilisant le paired- informations sur l'échantillon ou non. Cet ensemble comprend des gènes présentant le changement, l'expression la plus cohérente (plus de 2 fois) dans le cancer par rapport
tissus témoins, qui ont été considérés comme différentiellement gènes avec une grande fiabilité exprimés, issus de multiples tests statistiques. En revanche, l'ensemble des 452 gènes représentent un ensemble étendu. Nous avons constaté qu'il existe une tendance générale que le nombre des gènes exprimés de manière différentielle augmente en cancer de l'estomac, par rapport à un tissu normal, est plus mal différencié, comme le montre la figure 1. Cette observation est en accord avec notre connaissance générale que Lessing cancers différenciés ont tendance à avoir des gènes exprimés de manière différentielle plus et sont plus agressifs; l'exception pour WD, comme le montre la figure 1, pourrait refléter les petites tailles de la WD et les groupes MD.
| = 1, P
-value < 0,05) (voir les détails dans le tableau S2). Parmi ces gènes sont POF1B
, MET
, CEACAM6
, ZNF367
, GKN1
, LIPF
, SLC5A5
, MUC13
, CLDN1
, MMP7 et ATP4A
, qui sont tous connus pour être liés au cancer. La figure 2 montre quatre exemples avec soit des corrélations positives ou négatives. Parmi eux, MUC13
a été rapporté comme un bon marqueur pour le niveau de différenciation de la muqueuse gastro-intestinale [13]. Une expression accrue MUC13 a été trouvée pour induire des changements morphologiques, y compris la diffusion des cellules à travers l'interférence avec la fonction des molécules d'adhésion cellulaire [14]; ainsi, une expression accrue ainsi que la différenciation peut indiquer une meilleure adhésion cellule-cellule
B. Identification des signatures génétiques pour les classes de cancer
tissus témoins. De la même façon, un groupe de 198 gènes peut distinguer parmi les quatre niveaux différents de cancer et le groupe témoin en fonction de leur expression génique, ce qui donne lieu à 74,2% précision globale de classification. Les deux ensembles de gènes ont été choisis en fonction d'un vote à la majorité (au moins 70% de consistance) régime à partir des résultats de classification sur 500 ensembles échantillonnés au hasard parmi les ensembles d'échantillons 54, ainsi que leur signification de classement (voir Méthodes pour les détails).
pour WD; GKN2
pour MD; CSTC
pour PD; et un gène régulé à la baisse, RHOJ,
pour le groupe UD. Ces discriminateurs monogéniques montrent AUCs significatives, allant de 0,76 à 0,99, tandis que la précision globale de classification obtenus par 5 fois gamme de validation croisée de 70,0% à 97,0% pour les différents groupes. Une recherche ultérieure pour k
combinaisons -Gene (k = 2, 3, 4) pour chaque groupe de cancer en passant de manière exhaustive à travers toutes les combinaisons de k
groupes -Gene également identifiés.
pathologique
, MUC17
, OR9G1
, REEP3 et TMED6
sont des protéines membranaires , pour la plupart des récepteurs qui transduction des signaux extracellulaires. DEFA5
est un peptide microbicide censé être impliqué dans la défense de l'hôte qui est fortement exprimée dans l'iléon [16]. CPS1
, DES et TTN
sont impliqués dans plusieurs processus métaboliques, la fonction musculaire et la phase M du cycle cellulaire mitotique, respectivement. Nous pensons que ces gènes SIGNALISATION et immuno liés peuvent représenter l'anomalie précoce des cellules du tissu pendant l'oncogenèse en général.
, (la figure 4) de MFAP2 et PPA1, montrant cohérente en amont et en régulation, respectivement, ainsi que la progression du cancer.
tissus de référence correspondants. Nous avons constaté que la plupart sont impliquées dans les processus extracellulaires telles que l'adhérence focale, chorio jonction étanche, l'interaction des récepteurs des cytokines de cytokines et de l'interaction ECM-récepteur, la cascade d'activation du plasminogène, ainsi que les voies de signalisation, y compris Wnt et de signalisation d'intégrine, qui sont étroitement liés à la croissance cellulaire et le contrôle de la prolifération cellulaire. Recherche contre notre base de données interne (http://bioinfosrv1.bmb.uga.edu/DMarker/) qui comprend des ensembles de données de microréseaux publics de GEO [17], Oncomine [18] et SMD [19], couvrant plus de 53 maladies humaines, y compris le cancer, nous avons constaté que les profils d'expression différentielle de 15 gènes sont hautement spécifique au cancer gastrique, tels que GKN2, CLDN7, THY1, GIF et PGA4, alors que la plupart des autres sont en général à plusieurs types de cancer. Par exemple, les plus généraux comprennent quelques membres de la famille des gènes de collagène (COL1A2, COL3A1 et COL1A1), la molécule d'adhésion cellulaire liés antigène carcinoembryonnaire (CEACAM6), métalloprotéinases matricielles (MMP1, MMP7 et MMP12), la topoisomérase (TOP2A) et phosphoprotéine sécrétée (de SPP1).
, CLDN1 et DPT
, de ces gènes sont significativement différenciés dans toutes les classes ou les stades du cancer gastrique. Nous pouvons voir sur la figure 5A et 5B que les deux CLDN7 et CLDN1
sont fortement exprimé dans le cancer par rapport
échantillons de contrôle dans tous les grades et stades, avec une augmentation modérée dans les tissus cancéreux début, alors que DPT
a été régulée à la baisse dans tous ces groupes. Le modèle d'expression cohérente à travers tous les sous-groupes de cancer peut indiquer que ces gènes participent à de nombreuses grandes voies biologiques impliquées dans la formation du cancer et de la progression. Comme cela est bien connu, les deux protéines Claudin, claudine-1 et claudine-7, sont des protéines membranaires intégrales essentielle pour la formation des jonctions serrées, le maintien de l'adhésion de cellule à cellule et de régulation paracellulaire et le transport transcellulaire de solutés à travers les epitheliums humains et les endotheliums, qui sont différentiellement exprimés dans divers cancers tels que le cancer du col [20], le carcinome rénal [21] et un type intestinal du cancer gastrique [22]. Dermatopontin (
TPN) est une protéine de matrice extracellulaire servant d'une liaison de communication entre la surface de cellules de fibroblastes du derme et sa matrice extracellulaire. Son expression réduite a également été trouvée dans les deux léiomyomes et chéloïdes [23] utérins. ROC représenté sur la figure 5C indique que ces gènes pourraient éventuellement être utilisés comme marqueurs efficaces pour le diagnostic du cancer de l'estomac en général.
E. La vérification des signatures identifiées sur des ensembles de données publiques
et Takeno de jeux de données (voir matériels et Méthodes), afin de déterminer la généralité de ces signatures génétiques. Comme on le voit sur la figure 6, la répartition des différences d'expression entre les données et Kim ensemble de données est sensiblement concordant, ce qui indique que l'applicabilité générale de nos marqueurs identifiés. Sur les 19 et 12 gènes se chevauchant des grades-corrélés identifiés ci-dessus et la liste des gènes stade-corrélé, 10 et profils d'expression 5 montrent similaires dans les cancers de grades G1-2 /G3-4 et stades I-IV dans le les données Kim, respectivement, ce qui reflète une forte cohérence dans les profils d'expression de ces gènes entre les différents ensembles d'échantillons.
données et obtenu 78,0% de précision de classification sur la validation croisée de 5 fois en termes de distinction peu de cancers hautement différenciés. De même, les signatures à deux étages (10 groupes de gènes et 9 gènes) obtenus exactitudes respectives de 84,0% et de 76,0% par rapport à l'ensemble de données
Kim. La signature 198 gène n'a pas été vérifié depuis le Kim ensemble de données fournit seulement pliez changement au lieu de données d'expression premières.
expériences
Microarray Data Analysis
était inférieur à 4 (intensité du signal normalisé).
tissus de référence, comme suit. Pour chaque gène, K exp
, le nombre de paires de tissus cancer /référence dont l'expression-facteur de changement (FC) est plus grand que k
(par exemple k
= 2) a été examiné; si la P-valeur pour l'observé K exp
a été inférieur à 0,05, le gène a été considéré comme étant différentiellement exprimés dans la majorité des paires de tissus cancéreux et de référence (voir les informations à l'appui). Notre P-valeur calculée n'a pas été ajustée sur les tests de la multiples hypothèses afin d'éviter toute perte de gènes qui peuvent être potentiellement efficace dans l'étape de classification ultérieure.
Sélection génétique et classification
pour le i
fonction -ème, calculée en utilisant la formation mis seulement. Lorsqu'un gène est supprimé ou son poids w i est réduit à zéro, la variation de la fonction de coût J (i) est donnée par
. Le cas de correspond à la suppression de la i
gène -ème. Le changement de la fonction de coût indique la contribution du gène à la fonction de décision et sert comme un indicateur de classement du gène
.
données de biopuces publics du cancer gastrique
(GSE3438) et les (GSE15081)
Takeno ensembles de données. Le premier [34] comprend l'expression des gènes de 50 patients gastriques de cancer (de Corée) à différents stades et le niveau de différenciation, qui a été utilisé pour vérifier la cohérence de nos signatures identifiées. Les données Takeno [24] comprend 141 tissus de cancer gastrique primaire après chirurgie curative, avec des informations rechute péritonéale suivi. Ces ensembles de données fournissent le rapport de log2 normalisée de la tumeur et l'expression normale.
Informations complémentaires
Tableau S1.
Statistiques de 452 gènes qui sont exprimés de manière différentielle dans l'un des groupes de quatre grades, déterminé selon les critères suivants: les niveaux d'expression dans le cancer et l'exposition des tissus de contrôle correspondant de changement au moins 2 fois, et le seuil de signification statistique d'avoir ce niveau de changement d'expression est P
-value < 0,05
doi:. 10.1371 /journal.pone.0017819.s001
(XLSX)
Tableau S2.
99 gènes ont leur expression change parfaitement en corrélation avec les grades WD-MD-PD-UD (| CC
| = 1, p
-value < 0,05).
doi: 10.1371 /journal.pone.0017819.s002
(XLSX)
Tableau S3.
Liste des noms des gènes de la signature de 198 gènes, parmi lesquels 39 sont des gènes de qualité corrélés. CC:. Coefficient de corrélation
doi: 10.1371 /journal.pone.0017819.s003
(XLSX)
Tableau S4.
Liste des 40 gènes de signature qui se trouvent spécifiquement pour le groupe WD; 18, 20 et 255 gènes sont spécifiques au groupe MD, PD et UD, respectivement
doi:. 10.1371 /journal.pone.0017819.s004
(XLSX)
Tableau S5.
 Les femmes sont-elles plus susceptibles de développer un COVID long ?
Les femmes sont-elles plus susceptibles de développer un COVID long ?
 Des chercheurs manipulent des espèces bactériennes dans l'intestin à l'aide d'un régime alimentaire
Des chercheurs manipulent des espèces bactériennes dans l'intestin à l'aide d'un régime alimentaire
 Les antioxydants dans l'alimentation pourraient augmenter le risque de cancer de l'intestin,
Les antioxydants dans l'alimentation pourraient augmenter le risque de cancer de l'intestin,
 La maladie du côlon irritable augmente le risque de démence
La maladie du côlon irritable augmente le risque de démence
 Problèmes de croissance chez les nouveau-nés prématurés associés à des bactéries intestinales altérées
Problèmes de croissance chez les nouveau-nés prématurés associés à des bactéries intestinales altérées
 Le microbiote rural et urbain diffère dès le plus jeune âge,
Le microbiote rural et urbain diffère dès le plus jeune âge,
 La thérapie biologique peut réduire le risque de COVID-19 sévère
Une récente étude observationnelle menée par des chercheurs espagnols, actuellement disponible sur le medRxiv* serveur de préimpression, suggère que les patients atteints de maladies à médiation imm
La thérapie biologique peut réduire le risque de COVID-19 sévère
Une récente étude observationnelle menée par des chercheurs espagnols, actuellement disponible sur le medRxiv* serveur de préimpression, suggère que les patients atteints de maladies à médiation imm
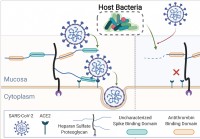 Le microbiome humain coupe les glycanes muqueux,
influençant linfection par le SRAS-CoV-2 Une équipe internationale de chercheurs a mené une étude montrant que les différences dans le microbiome humain peuvent influencer la capacité du coronavirus 2
Le microbiome humain coupe les glycanes muqueux,
influençant linfection par le SRAS-CoV-2 Une équipe internationale de chercheurs a mené une étude montrant que les différences dans le microbiome humain peuvent influencer la capacité du coronavirus 2
 Une étude suggère un lien entre l'utilisation de probiotiques et le « brouillard cérébral »
Une étude menée au Medical College of Georgia de lUniversité dAugusta a montré que la consommation de probiotiques peut entraîner une accumulation importante de bactéries de lintestin grêle qui entraî
Une étude suggère un lien entre l'utilisation de probiotiques et le « brouillard cérébral »
Une étude menée au Medical College of Georgia de lUniversité dAugusta a montré que la consommation de probiotiques peut entraîner une accumulation importante de bactéries de lintestin grêle qui entraî