Helicobacter pylori (H. pylori) Visto:. Moen EL, Wen S, Anwar T, Cross-Knorr S, brillante K, Birnbaum F, et al . (2012) regolazione della funzione RKIP da Helicobacter pylori Editor: Yoshio Yamaoka, Veterans Affairs Medical Center (111D), Stati Uniti d'America Ricevuto: 30 Dicembre 2011; Accettato: 24 Aprile 2012; Pubblicato: 25 maggio 2012 Copyright: © 2012 Moen et al. Questo è un articolo ad accesso libero distribuito sotto i termini della Creative Commons Attribution License, che permette l'uso senza restrizioni, la distribuzione e la riproduzione con qualsiasi mezzo, a condizione che l'autore originale e la fonte sono accreditati Finanziamento:. Questo lavoro è stato sostenuto dal National Institute of General Medical Sciences del National Institutes of Health sotto Premio Numero P20GM103421 (DC); il segmento precedente di questo progetto è stato sostenuto dal National Center for Research Resources (NCRR) sotto P20 RR 017.695 (DC), R01 CA111533 (SFM) e R21 CA133601 (JMS). I finanziatori avevano alcun ruolo nel disegno dello studio, la raccolta e l'analisi dei dati, la decisione di pubblicare, o preparazione del manoscritto Competere interessi:.. Gli autori hanno dichiarato che non esistono interessi in competizione Introduzione cancro gastrico è la quarta neoplasia più frequentemente diagnosticata in tutto il mondo. Nel 2007, circa un milione di nuovi casi di cancro gastrico che porta a circa 800.000 morti in tutto il mondo sono stati registrati, il che rende la seconda causa più comune di morte per cancro [1]. cancro gastrico è attualmente la settima causa di decessi per cancro negli Stati Uniti, con circa 21.500 nuovi casi diagnosticati nel 2011 (http://www.cancer.gov/cancertopics/types/stomach). Il, a forma di spirale batterio gram-negativo Helicobacter pylori (H. pylori) H. pylori proteine STAT sono costitutivamente espressi in diverse neoplasie, tra cui gastrica, della mammella, della testa e del collo, e tumori della prostata [13] - [16].. Su fosforilazione della tirosina 705 residui e acetilazione in lisina 685, dimerizza STAT3 ed entra nel nucleo dove funziona a trascrizionalmente regolare un'ampia gamma di geni [17], [18]. è stato dimostrato che l'attivazione costitutiva della proteina STAT3 per prevenire l'apoptosi e aumentare la proliferazione delle cellule e metastasi in un certo numero di tumori, tra cui il cancro gastrico [19], [20] Una delle caratteristiche di progressione del tumore gastrico è la acquisizione di fenotipi più invasive e migratorie durante la transizione epitelio-mesenchimale (EMT). Durante EMT, cellule epiteliali gastriche subiscono variazioni fenotipiche caratterizzato dalla perdita di molecole di adesione cellulare, in particolare la epiteliale caderina (E-caderina) [21]. Il fattore di trascrizione lumaca, una proteina zinc-finger, è stato caratterizzato in precedenza come un importante regolatore di EMT per la sua attivazione tramite Nuclear Factor kappa Beta (NF-kB) [22] e la successiva repressione di E-caderina in cellule tumorali epiteliali [ ,,,0],23], [24]. Inoltre, gli studi con il guadagno-di-funzione e la perdita-di-funzione approcci hanno identificato lumaca come un repressore della trascrizione RKIP nelle cellule di carcinoma della prostata metastatico [25]. RKIP è un membro della proteina fosfatidiletanolammina vincolante famiglia e un regolatore negativo della (chinasi extracellulare signal-regulated) ERK1 /2 [26], NF-kB [27] e GRK (G protein-coupled Receptor Kinase) [28] percorsi. RKIP svolge quindi un ruolo importante nella regolazione della sopravvivenza cellulare e apoptosi, oltre a potenziare l'efficacia di agenti chemioterapici [29]. RKIP è stato anche identificato come una proteina soppressore delle metastasi [30], e nei pazienti con adenocarcinoma gastrico esiste una correlazione positiva tra espressione RKIP e la sopravvivenza del paziente e una correlazione inversa tra l'espressione di STAT3 RKIP e [19]. espressione e la funzione RKIP possono essere regolate da modificazioni post-traslazionali. Ad esempio, la fosforilazione di RKIP dalla proteina chinasi C a serina-153 previene la capacità di RKIP di legarsi alla sua molecola bersaglio, inattivando così la funzione RKIP [31]. Inoltre, la repressione RKIP tramite metilazione del promotore può essere superato con inibitori della deacetilasi degli istoni metilazione e [25]. A causa dei ruoli importanti di RKIP, STAT3 e H. pylori Materiali e Metodi Reagenti Tutti i reagenti e sostanze chimiche sono stati acquistati da Sigma Chemical Co. (St. Louis, MO) a meno che diversamente specificato. MG-132 è stato acquistato dalla Calbiochem (Gibbstown, NJ) disciolto in DMSO e utilizzato ad una concentrazione di 10 mM. L'interleuchina-6 (IL-6) è stato acquistato da BD Biosciences (San Diego, CA). reagenti quantificazione della proteina sono stati ottenuti da Bio-Rad (Hercules, CA). reagenti chemiluminescente e secondario del mouse e coniglio perossidasi di rafano-coniugato per l'analisi Western Blot sono stati da GE Healthcare (Piscataway, NJ). L'actina-HRP, fosforilata-RKIP (pRKIP) e anticorpi STAT3 sono stati acquistati da Santa Cruz Biothechnology (Santa Cruz, CA). Gli anticorpi di STAT3 pS727 e pY705 e PARP sono stati acquistati da Cell Signaling Technology (Beverly, MA) e l'anticorpo per RKIP da Millipore, Billerica, MA. L'anticorpo di lumaca è stato acquistato da Abcam (Cambridge, MA). La linea cellulare umana carcinoma gastrico AGS (CRL-1739) è stato acquistato da American Type Culture Collection (Manasas , VA). cellule MKN28 sono stati donati dal Dr. Richard Peek, Vanderbilt University, Nashville, TN e sono stati acquistati da Riken Cell Bank, Ibaraki, in Giappone. I plasmidi di espressione per pcDNA3, c-myc STAT3, CMV-HA-RKIP (HA-RKIP) e CMV-HA vettore vuoto (EV) sono stati descritti [18], [26]. Il plasmide RKIP S153V è stato fornito dal Dr. Marsha Rosner, University of Chicago, Chicago, IL. tipo selvaggio H. pylori Trasfezione di cellule AGS cellule AGS sono state trasfettate utilizzando il reagente plasmide trasfezione GenJet (Signagen Laboratories, Gaithersburg, MD) secondo il protocollo del produttore per un formato 6-pozzetti. Quantitativi totali DNA tra 1 e 2 mg sono state trasfettate per campione. condizioni di trasfezione sono stati valutati e ottimizzati per l'analisi di cellule trasfettate con un Green Fluorescent Protein (GFP) esprimente RKIP plasmide. efficienze di trasfezione sono stati costantemente nel range 75-85%. proteine Estrazione e Western Blot analisi estratti cellulari totali e frazionamenti subcellulari sono stati preparati e immunoblotted come in precedenza [29], [32 descritta ]. concentrazioni di proteine sono state determinate utilizzando il BCA Protein Assay (Thermo Scientific). Densitometria di Western blot è stata eseguita secondo il protocollo indicato al seguente sito:. Http://lukemiller.org/journal/2007/ Realtime PCR Due mg di RNA è stato convertito in cDNA usando RevertAid First-Strand cDNA Synthesis Kit (Thermo Scientific). Quantitativa real-time PCR è stata effettuata utilizzando 2 × QIAgen QuantiFast SYBR Green I (Roche). I primer per Lumaca erano avanti: AGCTCTCTGAGGCCAAGGATCT, invertire: TGTGGCTTCGGATGTGCAT e beta-actina: in avanti: CTGGCACCACACCTTCTACAA, invertire: CAGCCTGGATAGCAACGTACA. I seguenti tempi profili tipici utilizzati erano per 40 cicli: una fase iniziale a 95 ° C per 10 minuti, seguito da 95 ° C per 15 s e 60 ° C per 1 min. Il livello di espressione relativo è stato calcolato con il metodo 2-ΔΔCT come descritto in precedenza [33]. Celle (2 × 10 5 cellule /pozzetto in 6 pozzetti) sono state trasfettate con 0,1 mcg (STAT3, RKIP) o 0,05 mg (NF-kB) di un plasmide giornalista che contiene sia il legame STAT3 SIE-frammento della regione del promotore del gene del mouse IRF1 (p2xSIE-Luc) o promotore regione RKIP più i plasmidi indicati come precedentemente descritto [18]. Circa 24 ore dopo la trasfezione, le cellule sono state trattate con il farmaco indicato o infettati con H. pylori L'apoptosi è stata quantificata in saggi separata mediante citometria di flusso e la frammentazione del DNA ELISA. Per citometria a flusso, la percentuale di cellule apoptotiche (sub-G O) è stata determinata mediante citometria a flusso delle cellule ioduro di propidio macchiato [29]. Citoplasmatica istone-associata frammentazione del DNA è stata misurata con il Cell Death Detection ELISA Inoltre kit (Roche, Indianapolis, IN) secondo le istruzioni del produttore. citoplasmatica istone-associata frammentazione del DNA è stata misurata con il Cell Death Detection ELISA più kit (Roche, Indianapolis, IN) secondo le istruzioni del produttore. Gli esperimenti sono stati ripetuti 3 volte e eseguiti in duplicato. Lentivirus costruisce. pLKO.1 puro resistenza lentiviral costruire RHS3979-97070798 e RHS3979-98492779 sono stati acquistati da Open Biosystems (Huntsville, AL). I costrutti contenevano un marcatore di selezione puromicina e sono state coltivate in Luria Broth contenente ampicillina a 37 ° C. Il brodo è stato centrifugato a 10.000 × g cellule imballaggio 293T sono state seminate in mezzi a basso antibiotico crescita (DMEM, il 10% del calore inattivato FBS, 0,1 × penicillina /Strepomycin /glutammina). Le cellule sono state incubate per 24 ore (37 ° C, 5% CO 2), o fino a quando non sono stati circa ~70% confluenti. I media sulle cellule imballaggio 293T è stato sostituito con supporti ad alta crescita contenente DMEM. Una miscela dei plasmidi trasfezione sono stati preparati come segue:. Imballaggio plasmide (ΔVpr.89), busta plasmide (VSV-G), tornante-pLKO.1 e vettore vuoto reagente FuGENE trasfezione è stata preparata in DMEM secondo le istruzioni del produttore. Brevemente, i 3 plasmidi sono stati aggiunti goccia a goccia alla Fugene e DMEM e misti. La miscela viene quindi lasciata incubare per 20-30 min. a temperatura ambiente. La miscela di trasfezione è stata poi accuratamente aggiunto alle cellule di confezionamento. Le cellule sono state incubate per circa 18 h. I media trasfezione è stata quindi scartata la mattina seguente e sostituito con supporti ad alta crescita. Le cellule sono state incubate per 24 h. containingand lentivirus contenente i media è stato raccolto ulteriore supporti ad alta crescita aggiunto. Le cellule sono state incubate per 24 ore e raccolte dai media. raccolta tipica era per 2-3 punti di tempo. Tutti raccolti virali sono stati raggruppati. cellule AGS sono state coltivate a circa il 50% di confluenza. I supernatanti virali sono stati aggiunti con polibrene. Dodici ore dopo l'infezione, i media virale è stato scartato e sostituito con i media virali e incubate per altri 12 h. I mezzi di comunicazione è stata scartata e sostituita con il mezzo F12 di Ham. Le cellule sono state incubate per 24 h. Le cellule sono state suddivise in mezzi di selezione contenenti di puromicina e lasciati incubare per 24 h. Tutti gli esperimenti di coltura cellulare sono stati ripetuti almeno 3 volte, se non diversamente indicato, e accoppiati t- test sono stati utilizzati per determinare la significatività statistica. Risultati H. pylori RKIP inibisce diversi percorsi di sopravvivenza delle cellule, comprese quelle mediate attraverso NF-kB e Jak /STAT [22]. Per chiarire l'effetto di H. pylori H. pylori La fosforilazione di RKIP sulla serina 153 dalla proteina chinasi C (PKC) abroga la sua capacità di legarsi a Raf e inibire a valle MAP chinasi segnalazione [31]. Abbiamo esaminato se la fosforilazione di RKIP da H. pylori IL-6 Induce fosforilazione di RKIP e H. pylori Poiché non vi è una relazione inversa tra RKIP e l'espressione di STAT3 in campioni di cancro gastrico [19], abbiamo valutato se la STAT3 e il suo regolatore chiave di IL-6 [17] colpisce espressione pRKIP. IL-6 trattamento alla dose di 25 o 50 ng /ml aumento dei livelli di proteine pRKIP 1,8 e 1,35 volte, rispettivamente, e l'espressione di proteine totali RKIP diminuita 0,8 e aumentato 1,05 volte, rispettivamente (Fig. 2A). La fosforilazione di RKIP in risposta a IL-6 era PKC-dipendente (dati non mostrati). Questi dati indicano che IL-6 può anche indurre la fosforilazione di RKIP in cellule gastriche che possono verificarsi, in parte, ad una via PKC-dipendente. Macrofagi citochine rilascio, tra IL-6 durante H. pylori Phosphorylated RKIP induce il proprio Trascrizione Abbiamo utilizzato un saggio luciferasi giornalista RKIP per studiare gli effetti di H. pylori H. pylori Per valutare il ruolo della specifica H. pylori Per verificare se la mutazione della serina 153 colpisce H. pylori H. pylori A causa aumento della trascrizione RKIP indotti da H. pylori Un altro meccanismo che potrebbe spiegare la mancanza di cambiamento nella proteina RKIP o l'espressione di mRNA sarebbe repressione trascrizionale di RKIP dopo H. pylori H. pylori Discussione gastrite cronica e alterato turnover cellulare indotta da H. pylori Il nostro studio descrive un altro meccanismo attraverso il quale H. pylori Cag-positivo H. pylori Infezioni con cagPAI-in possesso di ceppi di H. pylori Quali sono associati con una risposta infiammatoria più forte nello stomaco e posa un rischio maggiore di sviluppare ulcera peptica o cancro allo stomaco di ceppi privi dell'isola cag [36]. H. pylori
è un batterio spiraliforme gram-negativi che infetta più della metà della popolazione mondiale ed è una delle principali cause di adenocarcinoma gastrico . I meccanismi che legano H. pylori
infezione alla carcinogenesi gastrica non sono ben compresi. Nel presente studio, si segnala che la proteina inibitore della Raf-chinasi (RKIP) ha un ruolo nella induzione di apoptosi da H. pylori
nelle cellule epiteliali gastriche. saggi di macchia e trascrizione reporter luciferasi occidentali dimostrano che la patogenicità isola di H. pylori
fosforila rapidamente RKIP, che poi si localizza nel nucleo dove attiva la propria trascrizione e induce apoptosi. iperespressione forzata di RKIP migliora l'apoptosi in H. pylori
cellule -infected, mentre RKIP RNA inibizione sopprime l'induzione di apoptosi da H. pylori
infezione. Mentre inducendo la fosforilazione di RKIP, H. pylori
obiettivi contemporaneamente RKIP non fosforilata per la degradazione proteasoma-mediata. L'aumento dei RKIP trascrizione e la fosforilazione è abrogata dalla mutazione RKIP serina 153 a valina, a dimostrazione che la regolamentazione di attività RKIP da H. pylori
dipende residuo S153 di RKIP. Inoltre, H. pylori
infezione aumenta l'espressione di lumaca, un repressore trascrizionale di RKIP. I nostri risultati suggeriscono che H. pylori
utilizza una proteina soppressore del tumore, RKIP, per promuovere l'apoptosi nelle cellule di cancro gastrico
nel cancro gastrico. PLoS ONE 7 (5): e37819. doi: 10.1371 /journal.pone.0037819
infetta più della metà della popolazione mondiale ed è stata identificata come un fattore di rischio nella carcinogenesi gastrica [2]. L'Organizzazione Mondiale della Sanità e l'Agenzia Internazionale per la Ricerca sul Cancro designati come classe I cancerogena nel 1994 [3]. La nostra attuale comprensione della H. pylori
carcinogenesi indotta è che il batterio e la risposta infiammatoria cronica associata promuovere gastrica morte delle cellule epiteliali per apoptosi [4], con conseguente iper-proliferazione [5], e la produzione di radicali liberi [6] i quali contribuiscono ad un lenta e progressiva sequenza di cambiamenti della mucosa gastrica che infine favoriscono progressione verso il cancro. Questo modello è coerente con i rapporti che i polimorfismi del gene di citochine pro-infiammatorie che aumentano l'intensità della risposta infiammatoria sono legati ad un aumento del rischio di cancro gastrico [7].
aderisce strettamente alle cellule epiteliali gastriche e può indurre l'apoptosi direttamente [8]. La patogenicità isola CAG (gene citotossici-associato) (CAG PAI) di H. pylori
è un segmento 40 kB di DNA che contiene geni che codificano per i componenti di un sistema di secrezione batterica tipo IV [9]. All'interno di questa regione è il cagA
gene che codifica per CagA, una proteina immunodominante di 121-145 kDa [9]. H. pylori
ceppi che possiedono e che esprimono il cag PAI sono più spesso associati con ulcera peptica e cancro gastrico nelle popolazioni occidentali rispetto ai ceppi che non [9]. Al momento della sua iniezione attraverso il sistema di secrezione IV tipo in ospitanti gastriche cellule epiteliali, CagA può successivamente diventare fosforilata da Src famiglie tirosin chinasi al suo C-terminale [10], che porta CagA di legare e attivare SHP2 e segnale attraverso ERK [11]. È importante sottolineare che, CagA è anche responsabile per l'attivazione del trasduttore di segnale e attivatore di trascrizione 3 (STAT3) in vitro
e in vivo
[12], anche se questo potrebbe non essere necessariamente dipendere CagA fosforilazione [11]
.
nella patogenesi del cancro gastrico, abbiamo studiato se H. pylori
segnali attraverso RKIP. I nostri studi suggeriscono che una complessa interazione tra H. di pylori cagPAI
, RKIP, STAT3, e lumaca agisce per dysregulate gastrica apoptosi delle cellule epiteliali modulando la funzione RKIP, un meccanismo che definisce un ruolo centrale per RKIP in H. pylori
-associated carcinogenesi gastrica.
Celle e plasmidi
H. pylori
Ceppi e Cultura condizioni
ceppi o isogenico H. pylori
mutanti sono stati co-coltura con le AGS o linee cellulari gastrico MKN come descritto in precedenza [32] ad una molteplicità di infezione (MOI) di 100:1 in tutta esperimento se non indicato diversamente.
STAT3 e RKIP reporter luciferasi Assays
durante la notte o non trattata. L'attività luciferasi nel surnatante citosolico è stata valutata utilizzando il reporter luciferasi Assay (Promega) e misurata con un luminometro per stimare l'attività trascrizionale [18].
Apoptosis Saggi
Knockdown Lentivirus-mediata di RKIP
per 10 min. e il surnatante eliminato. DNA da pellet è stato purificato utilizzando il kit QIAGEN plasmidi Inoltre Maxi.
produzione Lentivirus.
infezione Lentivirus delle cellule AGS.
Metodi statistici
infezione aumenta la fosforilazione di RKIP
infezione RKIP in cellule gastriche, cellule AGS sono stati infettati con H. pylori
e raccolto 2 ore e 6 ore più tardi. Come mostrato in Fig. 1A, i livelli di RKIP fosforilata (pRKIP) sono stati elevati dopo 2 ore (3.126 volte) e 6 ore (2,9 volte) dopo H. pylori
infezione mentre l'espressione di proteine totali RKIP aumentato di 1,4 volte dopo 2 ore e 1,75 volte dopo 6 ore di H. pylori
infezione. Risultati simili sono stati ottenuti anche con cellule di cancro gastrico MKN (vedi Figura S1).
indotta fosforilazione di RKIP è PKC-dipendente
era PKC-dipendente. cellule AGS sono state infettate con H. pylori network per 6 h, in presenza o assenza di 40 pM Bisindolylmaleimide (Bis, un inibitore PKC). I nostri risultati indicano che i livelli di RKIP fosforilata è stata inibita 3,9 volte e RKIP 1,36 volte dopo H. pylori
infezione in presenza dell'inibitore PKC, suggerendo che la fosforilazione RKIP da H. pylori
coinvolge, ma potrebbe non essere del tutto dipendente, la via PKC-regolato (Fig. 1B).
si attiva STAT3
infezione [34] che porta all'attivazione STAT3 [17]. Per studiare gli effetti di H. pylori
infezione l'attivazione di STAT3, cellule AGS sono stati transitoriamente trasfettate con un IRF-1 giornalista costrutto [18] e co-coltura con H. pylori
al range indicato di molteplicità di infezione (MOI) per 24 h. I nostri risultati hanno dimostrato che ad una MOI tra 10-200:1, H . pylori
è stato in grado di indurre STAT3 trascrizione (Fig. 2C) e STAT3 pY705 fosforilazione (Fig. 2B) entro 6 ore di infezione. Abbiamo poi determinato se l'IL-6 potrebbe anche stimolare la trascrizione STAT3 in cellule AGS. cellule AGS sono state transitoriamente trasfettate con IRF-1 e con EV e o c-myc-tag STAT3 e poi dopo 24 h cellule trattati con IL-6 (50 ng /ml) o di co-coltura con H. pylori
a MOI di 100:1. I risultati, rappresentati in Fig. 2D, dimostrano che l'IL-6 (p < 0,0003) e H. pylori
(p < 0,0005) erano ciascuna in grado di stimolare in modo significativo la trascrizione STAT3, un effetto che è stato migliorato quando le cellule AGS sono state trasfettate con STAT3 e infettati con H. pylori
(p < ,0000,023 mila). La valorizzazione di attivazione di STAT3 è risultata significativamente aumentata quando le cellule AGS sono stati co-trattamento con IL-6 e H. pylori
rispetto al trattamento con IL-6 (p < 0,000,028 mila) o H. pylori
(p < 0,0003). sola
infezione RKIP attività trascrizionale. H. pylori
significativamente aumentata trascrizione RKIP (p < 0,002) con un aumento di oltre 10 volte si verificano con RKIP sovraespressione e un aumento maggiore di 16 volte con la combinazione di H. pylori
e RKIP (p < 0,0003) (Fig 3A.) rispetto alle cellule non trattate AGS trasfettate con vettore vuoto. C'è stato un aumento significativo (p < 0,0001) nella trascrizione RKIP con H. pylori
infezione e RKIP sovraespressione rispetto alle cellule trasfettate con RKIP senza infezione (Fig. 3A). Abbiamo ripetuto questi esperimenti in presenza di Bis di inibire l'attività PKC per determinare l'aumento RKIP trascrizione era dovuta alla fosforilazione. In presenza dell'inibitore PKC, H. pylori
aumentato RKIP trascrizione e RKIP sovraespressione portato anche nella valorizzazione di attività RKIP promotore. Bis diminuita RKIP trascrizione indotto da RKIP sovraespressione e H. pylori
infezione maggiore di 4 volte, rispetto alle cellule in con RKIP sovraespressione e suggerisce che questi effetti sono stati dipende RKIP fosforilazione (Fig. 3A). Abbiamo esaminato la localizzazione di RKIP dopo H. pylori
infezione. Immunoblotting subcellulari AGS cellule frazioni hanno dimostrato che pRKIP è localizzata nel nucleo mentre RKIP rimane principalmente nel citoplasma dopo l'infezione, (Fig. 3B). Insieme, questi dati implicano che H. pylori
può promuovere la traslocazione di pRKIP nel nucleo dove può attivare la trascrizione RKIP.
indotta RKIP fosforilazione Dipende H. cag di pylori
patogenicità Island e RKIP serina 153
fattori nella fosforilazione di RKIP, wild type H. pylori
e mutanti isogenici manca l'intero cag PAI o
il OIPA
gene erano co-coltura con cellule AGS per 6 ore. Il H. pylori
mutante manca la cag PAI
non era in grado di indurre RKIP fosforilazione, mentre il tipo macchia selvaggio e il OIPA
mutante fortemente indotta RKIP fosforilazione (Fig. 4A). La stessa tendenza è stata osservata su STAT3 pY705. Questi risultati suggeriscono che i geni all'interno di H. di pylori
cagPAI sono necessari per l'induzione di RKIP e STAT3 fosforilazione.
mediata fosforilazione RKIP e l'attivazione della trascrizione, le cellule sono state AGS trasfettate con un RKIP costruire in cui serina è stato sostituito con valina alla posizione 153 (S153V) e poi co-coltura con H. pylori
. Ancora una volta abbiamo osservato un maggiore di aumento di 16 volte della attività del promotore RKIP in cellule con RKIP sovraespressione e H. pylori
infezione (p < 0,0005). Tuttavia, nelle cellule trasfettate con RKIP S153V prima di H. pylori
infezione, c'è stata una riduzione di 2,5 volte l'attività trascrizionale (p < 0,0003) rispetto al H. pylori
infezione nel wild-type RKIP overexpressing cellule (Fig. 4C). Inoltre, la sovraespressione di S153V RKIP inibito H. pylori
mediata RKIP fosforilazione (Fig. 4B). Presi insieme, questi risultati indicano che la fosforilazione e trascrizionale attivazione di RKIP dipende H.
pylori cagPAI e anche sulla fosforilazione di RKIP a S153.
Risultati infezione nel RKIP degrado e l'induzione di lumaca
infezione non è stata associata ad un aumento dell'espressione proteica RKIP totale stato stazionario, abbiamo esaminato se H. pylori
potrebbe contemporaneamente aumentare il tasso di degradazione delle proteine RKIP attraverso la degradazione proteasoma-mediata, come suggerito in precedenza [35]. MG132 ha aumentato i livelli di proteina RKIP in presenza o assenza di H. pylori
infezioni, in linea con H. pylori
aumento del proteasoma RKIP degrado (Fig. 5A).
infezione. Lumaca è un fattore di trascrizione che gioca un ruolo importante in EMT [23], oltre ad essere un noto repressore trascrizionale RKIP nelle cellule di cancro della prostata [25]. Per studiare gli effetti di H. pylori
infezione sull'espressione di lumaca e RKIP, le cellule AGS sono stati co-coltura con H. pylori
ad una MOI di 100. lumaca mRNA espressione è stata fortemente indotta dopo 2-4 ore di infezione (Fig. 5D) analisi Western Blot ha indicato che H. pylori
infezione ha portato in un tempo e aumento dose-dipendente dei livelli di proteina Lumaca (Fig. 5B /C). Questo risultato non è coerente con i nostri dati sulla trascrizione RKIP dopo H. pylori
infezione (Fig. 3) e suggerisce che H. pylori
infezione può nell'induzione della proteina (s), che abolisce l'effetto di lumaca sulla trascrizione RKIP. Attualmente stiamo studiando questa possibilità con l'analisi Spettrometria di Massa utilizzando cellule parentali e RKIP atterramento (Fig. 6).
RKIP Migliora H. pylori
mediata apoptosi
induce l'apoptosi delle cellule epiteliali gastriche [8]. Dal momento che RKIP può promuovere l'apoptosi [29], abbiamo esaminato se l'induzione di pRKIP dopo H. pylori
infezione, potrebbe essere responsabile di H. pylori
apoptosi indotta. cellule AGS sono state trasfettate con RKIP o un vettore vuoto, infettati con H. pylori
per 16 ore e l'apoptosi valutati tramite PARP scissione citometria a flusso e la frammentazione del DNA. In alcuni esperimenti RKIP è stata inibita da lentivirus-mediata RKIP atterramento. Come mostrato in Fig. 6A, H
. pylori
indotto la scissione di PARP, un effetto che è stato aumentato di espressione ectopica di RKIP. Citometria a flusso analisi ha indicato che H. pylori
infezione ha determinato un aumento di circa 4 volte l'apoptosi (p < 0,0008), RKIP sovraespressione, un aumento di 3 volte (p < 0,003) e la combinazione di un aumento di 6 volte (p < 0,0005) rispetto ai cellule AGS non trattate (Fig. 6B). Nel saggio ELISA frammentazione del DNA-based, attività apoptotica aumentata: 2,5 volte (p < 0,000,063 mila) nelle cellule infettate con H. pylori
; 1,8 volte (p < 0,006) nelle cellule trasfettate con RKIP; e 3,5 volte (p < 0,0007) con la combinazione (Fig 6C.). Per determinare se RKIP è stato responsabile per H. pylori
apoptosi mediata, abbiamo soppresso l'espressione RKIP usando l'inibizione di RNA lentivirus-mediata e osservata una riduzione dei livelli di proteina RKIP da analisi Western Blot che dimostra la riduzione del RKIP a non trattati e H. pylori infettato cellule AGS (Fig. 6D). Nella nostra analisi della frammentazione del DNA, in cellule AGS parentali H. pylori
infezione ha determinato un aumento di 2 volte (p < 0,0007) in apoptosi (Fig 6E.). Nei RKIP atterramento cellule AGS, H. pylori
infezione ha determinato un aumento di 1,5 volte in apoptosi (p < 0,003) (Fig. 6E). La riduzione dell'apoptosi tra le cellule AGS parentali e RKIP atterramento era statisticamente significativa (p < 0,0006). Questi risultati indicano che RKIP è necessario per H. pylori
apoptosi mediata.
infezione promuovere lo sviluppo di adenocarcinoma gastrico distale [36]. H. pylori
può regolare l'apoptosi epiteliale gastrica attraverso diversi meccanismi. Ad esempio, dopo l'infezione e l'adesione alle cellule epiteliali gastriche, il sistema di secrezione cag serve a modificare intracellulare trasduzione del segnale conseguente attivazione di NF-kB. NF-kB può trasloca al nucleo per attivare la trascrizione dei geni pro-apoptotici [37]. H. pylori
può anche indurre apoptosi, aumentando l'espressione di FAS e il suo ligando (FASL) che porta alla attivazione della via estrinseca dell'apoptosi [38]. Paradossalmente, H. pylori
può anche attivare percorsi che downregulate apoptosi [39], specialmente in ritardo nel corso di infezione cronica [40]. Questa risposta adattativa delle cellule epiteliali di resistere apoptosi in H cronica. pylori
infezione può contribuire a H. pylori
carcinogenesi gastrica indotta [41]. La risposta apoptotica delle cellule epiteliali gastriche a H. pylori
dipende anche da fattori di virulenza specifici per ceppo. Ad esempio, l'infezione da ceppi CAG PAI-positivo può indurre l'apoptosi più rapidamente di cag PAI-negativi ceppi [42]. Il H. pylori vacA
prodotto del gene stimola la via apoptotica intrinseca che porta al rilascio mitocondriale di citocromo c, e caspasi-3 attivazione [43]. apoptosi indotta da VacA è associata con una riduzione di STAT3 porta alla sottoregolazione di Bcl-2 e Bcl-X L [43]. In un altro studio, è stato dimostrato che H. pylori
induce apoptosi da un percorso che coinvolge l'induzione sequenziale di apicale attività di caspasi-8, le proteine pro-apoptotici Bad e Bid, l'attività della caspasi-9, e effettori caspasi-3 attività [44].
infezione può promuovere l'apoptosi nelle cellule di cancro gastrico, in particolare attraverso la promozione di RKIP fosforilazione. La capacità di RKIP di inibire la segnalazione Raf /MAPK [26], [27] e di promuovere l'apoptosi è stata ben documentata [29]. L'interazione di tesi percorsi e livelli di espressione RKIP è stato implicato in molti passi della formazione del tumore e /o la progressione [30]. Inoltre, l'iperespressione di risultati RKIP nella inibizione di metastasi e invasività in vari modelli tumorali [45] - [48]. Il meccanismo alla base dell'espressione differenziale delle pRKIP e RKIP non è noto. Ci aspettavamo che i livelli relativamente elevati di pRKIP dopo l'infezione potrebbero correlare con i livelli più bassi RKIP. Tuttavia, abbiamo scoperto che H. pylori
infezione ha portato alla degradazione della proteina RKIP, eventualmente permettendo MAPK segnalazione e apoptosi induzione cancro gastrico dopo H. pylori
infezione. PKC-mediata RKIP fosforilazione può interrompere la capacità di RKIP di legarsi a Raf e inibire la segnalazione MAPK [31], tuttavia, ci non sono stati in precedenza alcun rapporto sul ruolo del pRKIP nella regolazione dell'apoptosi. Precedenti studi del nostro laboratorio hanno dimostrato che i risultati RKIP iperespressione nel attivazione diretta di pro-caspasi 8 [29]. Sebbene i risultati phosphoryation in relocalization nucleare, seguita dall'attivazione RKIP propria trascrizione, i livelli di proteina RKIP non aumentano. Ciò suggerisce un diverso meccanismo di regolazione RKIP quello che è stato riportato in precedenza. Attualmente stiamo esaminando il meccanismo con cui pRKIP innesca l'apoptosi nelle cellule di cancro gastrico dopo H. pylori
infezione.
upregulate significativamente i geni EMT-associata Chiocciola, Lumaca e vimentina in associazione con l'induzione di MMP-7, suggerendo un ruolo per queste proteine nello sviluppo del cancro gastrico [49]. Nel nostro studio abbiamo osservato la rapida induzione di proteine pRKIP dopo H. pylori
infezione, e un aumento della trascrizione RKIP e una sovraregolazione della lumaca mRNA e l'espressione della proteina. Anche se lumaca è stato identificato come un repressore trascrizionale di RKIP [25], il nostro studio indica che probabilmente ha alcun effetto sulla forma fosforilata di RKIP, dal momento che dopo l'infezione non abbiamo osservato una repressione della trascrizione RKIP.
induce una risposta infiammatoria intensa e localmente elevati livelli di diverse citochine tra cui l'interleuchina 6 (IL-6) [34].
 La dieta a basso contenuto di carboidrati fermentati migliora la qualità della vita dei pazienti con IBD
La dieta a basso contenuto di carboidrati fermentati migliora la qualità della vita dei pazienti con IBD
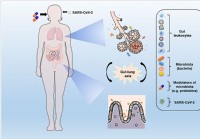 La modulazione del microbiota e il ripristino dell'eubiosi potrebbero aiutare a frenare le complicanze del COVID-19
La modulazione del microbiota e il ripristino dell'eubiosi potrebbero aiutare a frenare le complicanze del COVID-19
 Il microbioma polmonare prevede la gravità della malattia da COVID-19
Il microbioma polmonare prevede la gravità della malattia da COVID-19
 Le cellule immunitarie intestinali potrebbero essere responsabili dei cambiamenti del metabolismo secondo uno studio
Le cellule immunitarie intestinali potrebbero essere responsabili dei cambiamenti del metabolismo secondo uno studio
 Permeabilità intestinale e volo spaziale:il meccanismo rivelato
Permeabilità intestinale e volo spaziale:il meccanismo rivelato
 Una nuova strategia può rafforzare la comunicazione intestino-cervello
Una nuova strategia può rafforzare la comunicazione intestino-cervello
 Il pH acido migliora l'infezione da SARS-CoV-2 aumentando il recettore ACE2
La pandemia di coronavirus in corso del 2019 (COVID-19) causata da un nuovo coronavirus, vale a dire, sindrome respiratoria acuta grave coronavirus 2 (SARS-CoV-2), ha causato più di 4,6 milioni di vit
Il pH acido migliora l'infezione da SARS-CoV-2 aumentando il recettore ACE2
La pandemia di coronavirus in corso del 2019 (COVID-19) causata da un nuovo coronavirus, vale a dire, sindrome respiratoria acuta grave coronavirus 2 (SARS-CoV-2), ha causato più di 4,6 milioni di vit
 Lo studio rivela gli effetti antivirali della curcumina
curcumina, un composto naturale che si trova nella spezia curcuma, potrebbe aiutare a eliminare alcuni virus, la ricerca ha trovato. Uno studio pubblicato su Journal of General Virology hanno dimo
Lo studio rivela gli effetti antivirali della curcumina
curcumina, un composto naturale che si trova nella spezia curcuma, potrebbe aiutare a eliminare alcuni virus, la ricerca ha trovato. Uno studio pubblicato su Journal of General Virology hanno dimo
 I paesi con popolazioni più anziane hanno infezioni e decessi da SARS-CoV-2 più elevati,
dice studio A più di un anno dallinizio della pandemia di coronavirus 2019 (COVID-19), causata dalla sindrome respiratoria acuta grave coronavirus 2 (SARS-CoV-2), la differenza nella gravità della sin
I paesi con popolazioni più anziane hanno infezioni e decessi da SARS-CoV-2 più elevati,
dice studio A più di un anno dallinizio della pandemia di coronavirus 2019 (COVID-19), causata dalla sindrome respiratoria acuta grave coronavirus 2 (SARS-CoV-2), la differenza nella gravità della sin