stomaco Alla scoperta della storia umana dai batteri dello stomaco
astratti
recenti analisi di agenti patogeni umani hanno rivelato che le loro storie evolutive sono congruenti con il modello ipotizzato delle migrazioni delle popolazioni umane antiche e moderne. alberi filogenetici di ceppi del batterio Helicobacter pylori
e la polioma virus JC tratto da geograficamente diversi gruppi di esseri umani correlano strettamente con le relazioni delle popolazioni in cui si trovano.
Charles Darwin ha riconosciuto che la distribuzione e la forma di parassiti era evolutivamente significativo. Egli ha osservato, per esempio, che "... il pediculi [pidocchi] raccolti in diversi paesi dei differenti razze umane ... differiscono, non solo a colori, ma nella struttura dei loro artigli e degli arti. In ogni caso che molti esemplari sono stati ottenuti differenze erano costanti "[1]. Più recentemente, diversi gruppi di ricerca [2-7] hanno trovato correlazioni interessanti tra le relazioni evolutive tra i vari ceppi batterici e virali ospitati da esseri umani e il modello di migrazioni di esseri umani moderni di tutto il mondo.
Un caso particolarmente interessante è quella di Helicobacter pylori
, un batterio Gram-negativi associati a gastrite, ulcera peptica e cancro gastrico che possono infettare fino alla metà di tutti gli esseri umani [8]. La scoperta che una infezione batterica potrebbe portare a malattie croniche che erano considerate [8] era un esempio lampante del fatto che le malattie infettive non sono stati ancora conquistato. L'epidemia continua la sindrome da immunodeficienza acquisita (AIDS), focolai di Ebola in Africa centrale, e la diffusione attuale del virus del Nilo occidentale negli Stati Uniti e della sindrome respiratoria acuta grave (SARS) da Asia forniscono la prova della pervasività e conseguenze per la salute di agenti infettivi anche in età di vaccinazione e antimicrobica e terapie antivirali. Molte malattie infettive si pensa che siano sorti in concomitanza con lo sviluppo dell'agricoltura e l'aumento della vita urbana. Se, invece, le relazioni molti patogeni 'con gli esseri umani sono molto più antiche, non sarebbe sorprendente trovare associazioni evolutive più profondi tra gli esseri umani e la loro microbica e invasori virali.
La storia evolutiva di H. pylori
può fornire un esempio della coevoluzione di un batterio e il suo ospite solo conosciuta. Il H. pylori
genoma è relativamente piccolo a 1,67 megabasi, con un complemento minimo di geni metabolici [9]. Variazione tra H. pylori
isolati da persone diverse o anche da una sola persona è grande, portando a impronte digitali uniche per quasi ogni isolare finora digitato. I geni codificanti non sono molto diverse, comunque: maggior parte della variazione avviene nella terza posizione di base entro codoni o tramite inversioni o translo-cationi, lasciando le sequenze di aminoacidi codificati relativamente simili [2]. Questa conservazione della sequenza di amminoacidi è una fortuna per la ricerca del vaccino, come un vaccino è probabile che sia efficace su molti ceppi. Più interessante è il fatto che H. pylori
ha un tasso estremamente elevato di ricombinazione, superiori infatti quella di qualsiasi altro organismo caratterizzato finora [3, 10].
Normalmente, un alto tasso di ricombinazione sarebbe fare inferire storia evolutiva di un organismo molto difficile, in quanto le informazioni circa l'origine di ogni mutazione sarebbe perso come si diffonde in tutta una popolazione. Ma insieme con le modalità di trasmissione di H. pylori
, questo estremamente alto tasso di ricombinazione può infatti fare inferenze evolutive più facile. Diversi studi suggeriscono fortemente che H. pylori
è di solito trasmessa all'interno delle famiglie, in genere da madre a figlio [11, 12]. Così, la trasmissione di H. pylori
in qualche modo imita quella del DNA mitocondriale materno trasmessa [13]. Poiché il DNA mitocondriale viene trasmesso solo da un genitore (madre) e non ricombinano, ha dimostrato di essere un sistema genetico ideale per inferire storia evolutiva umana (vedi sotto) [14, 15]. Se H. pylori
è infatti prevalentemente materna trasmessa, nuovi ceppi in genere non infettare una persona durante la loro vita; insieme con l'elevato tasso di ricombinazione, ciò significherebbe che le mutazioni che si accumulano all'interno della popolazione di batteri nello stomaco di un individuo saranno relativamente omogenea. Ciò dovrebbe tradursi in uno sciame di ceppi che sono strettamente legati gli uni agli altri, che contiene molte delle mutazioni che si sono verificati nei singoli batteri. Gli sciami si trovano in diverse persone saranno così essere più diversi tra loro che se ci fosse meno ricombinazione.
Malattie infettive La maggior parte diffondono rapidamente in tutto il mondo e ceppi provenienti da diverse regioni sono relativamente simili, ma il campionamento iniziale di H. pylori
da persone provenienti da diverse regioni del mondo ha rivelato piuttosto forte partizionamento geografica in Asia H. pylori
tipi europei e [2-4]. Recentemente, Falush e colleghi [5] hanno esaminato questo partizionamento in modo più dettagliato. Dopo il sequenziamento otto geni - per un totale di 3.850 nucleotidi - in 370 ceppi provenienti da 27 popolazioni umane, hanno trovato 1.418 posizioni nucleotidiche polimorfici. Hanno poi applicato un nuovo strumento di analisi, STRUTTURA [16], che è stato sviluppato per dedurre la struttura genetica umana a partire dai dati genotipo multilocus. Il programma utilizza metodi bayesiani per identificare sottogruppi con frequenze alleliche distintivi, e gruppi di sottogruppi, anche in presenza di ricombinazione [16]. Quando questa tecnica è stata applicata alla H. pylori
sequenze, quattro gruppi principali sono stati trovati - due dall'Africa e uno ciascuno da Europa e Asia (Figura 1a) [5]. Figura 1 Le relazioni tra le popolazioni umane, come calcolato da H. pylori, ha trovato nello stomaco e dai dati del DNA mitocondriale. (A) I rapporti tra sottopopolazioni moderne di H. pylori
[5]. Ogni sottopopolazione è rappresentata da un cerchio con un diametro proporzionale alla diversità genetica all'interno di esso. I centri dei cerchi sono uniti da un albero filogenetico che mostra le relazioni tra le quattro sottopopolazioni. I batteri in ogni sottopopolazione si trovano prevalentemente nelle persone che provengono dalle regioni indicate. (B) Un albero filogenetico-livello di popolazione del H. pylori
sottopopolazioni geografiche mostrato in (a). (C) Una rete di popolazioni umane derivate da DNA mitocondriale [14] mediana-adesione. Tale rete mostra potenziali relazioni evolutive alternative tra cluster. Ogni cerchio rappresenta un gruppo di tipi mitocondriali di diametro proporzionale alla frequenza di questo tipo all'interno delle sottopopolazioni. Tutte le popolazioni non africane sono derivati da una stirpe africana; la rete di relazioni all'interno di questo lignaggio è ingrandita (in alto). (A, b) adattato da [5]; (C) adattato da [14]
Ogni cluster trovata da Falush e colleghi [5] potrebbe essere diviso in sottogruppi.; per esempio, cluster 'Africa 1' potrebbe essere suddiviso ulteriormente in Occidente e sottocluster sudafricani, e il cluster-est asiatico potrebbe essere diviso in orientale, Amerind e sottocluster Maori. Il partizionamento geografica entro i 200 ceppi europei è stato particolarmente complicato, presumibilmente perché numerosi gruppi hanno spazzato avanti e indietro in tutta Europa nel corso degli ultimi millenni. ceppi europei anche di tanto in tanto apparivano nelle Americhe, in Australia, e tra i sudafricani, presumibilmente riflettendo conquista coloniale.
Le relazioni filogenetiche di questi cluster (Figura 1B) e delle loro suddivisioni [5] mostrano un modello simile a quello ottenuto con il DNA mitocondriale variazione (Figura 1c) [14, 15]. Il moderno pool genetico umano, come si evince dal DNA mitocondriale e corroborata da studi del cromosoma Y, si pensa di aver avuto un'origine africana circa 150.000-200.000 anni fa [14, 15, 17, 18]. La popolazione umana originale poi diffuso e diversificato in tutta l'Africa per quasi 100.000 anni, prima di espandersi in Asia occidentale e in Europa e in Asia meridionale e orientale circa 50.000-60.000 anni fa, sostituendo le popolazioni arcaiche esistenti di esseri umani in queste regioni. migrazioni successiva diffusione in Australia da 40.000 anni fa, poi le isole del Pacifico, e più tardi in America del Nord, circa 15.000 anni fa (figura 2) [14, 15, 17, 18]. La notevole somiglianza tra questa visione della storia umana e i risultati degli studi di H. pylori
hanno portato Falush e colleghi [5] così come gli altri [2] per concludere che H. pylori
evoluzione ha seguito il percorso della moderna espansione umana e la migrazione. Questo lavoro fornisce così un altro tipo di dati per l'analisi dell'evoluzione umana e della migrazione, indipendentemente dal DNA mitocondriale e cromosomi Y, che saranno utili per ulteriori studi. Sfortunatamente, tuttavia, stimando la divergenza risale H. pylori
è particolarmente difficile, a causa del tasso estremamente elevato di ricombinazione [10]. tecniche di analisi aggiuntive campionamento e può essere richiesto di testare ulteriormente l'ipotesi migratoria. Figura 2 Una mappa del modello di espansione e migrazione dei moderni umani in tutto il mondo, derivate da studi di DNA mitocondriale e cromosomi Y [14, 15, 17, 18]. I numeri indicano il tempo approssimativo (in anni prima del presente) quando gli esseri umani moderni prima apparizione nella regione indicata
. Altri agenti patogeni sono stati anche proposti per seguire storie evolutive simili ai loro ospiti. Uno degli esempi più interessanti, anche se con un host non umano, è quella di afidi, un batterio trovato al loro interno, e due plasmidi associati con il batterio. Funk et al.
[19] ha rilevato che i filogenesi intraspecifrc dedotto di questi quattro genomi erano completamente congruenti. Tornando agli agenti patogeni umani, il virus JC poliomavirus umano (JCV) può essere suddiviso in genotipi che corrispondono alle grandi masse continentali [6]. Come H. pylori
, JCV - che può causare leucoencefalopatia multifocale progressiva (perdita di myelination nel sistema nervoso centrale) - è molto diffusa tra gli esseri umani come risultato di trasmissione familiare. Un totale di 12 sottotipi conosciuti sono stati definiti, con europei, africani, asiatici e le distribuzioni [7]. Anche se inferenze diretti di origine africana per JCV sono problematici perché non c'è outgroup adatto con cui sradicare l'albero filogenetico, quando un origine africana si presume, una storia evolutiva ragionevole può ipotizzare (Figura 3) [7]. Come da H. pylori
, inferire divergenza molecolare risale è attualmente problematico per JCV. Ulteriori indagini nella storia evolutiva di questi agenti patogeni umani è quindi necessario. Figura 3 I rapporti di polyoma umano virus JC (JCV) sottotipi riscontrati nell'uomo da diverse parti del mondo [7]. Le lettere si riferiscono ai singoli sottotipi. (A) Il modello ipotizzato di diffusione del JCV sottotipi attraverso il mondo (ad esclusione delle Americhe); (B) un filogenesi dedotto di sottotipi JCV, assumendo un origine africana per il virus. Adattato da [7].
Lo studio dei modelli di evoluzione dei patogeni umani possono in ultima analisi, fornire ulteriori elementi di prova non solo per la loro storia, ma anche di evoluzione umana e la storia. Questo sarà particolarmente vero per gli agenti patogeni, come H. pylori
che hanno una modalità prevalentemente madre-bambino di trasmissione di informazioni, che mimano l'evoluzione del DNA mitocondriale. H. pylori
s 'ruolo causale in diverse condizioni croniche dello stomaco è solo uno dei molti esempi attuali del ruolo nota o sospetta di un agente infettivo che porta a malattie croniche. I batteri sono sospettati di essere coinvolti nello sviluppo di arteriosclerosi, ictus, e la malattia di Crohn, mentre i virus sono noti per causare l'AIDS e le varie forme di epatite cronica. Il cancro della cervice, carcinoma epatocellulare, linfoma di Burkitt, sarcoma di Kaposi, e forse diabete mellito sono anche noto o sospettato di essere di origine virale. Se alcune delle malattie croniche onnipresenti dimostrare di essere di origine batterica o virale, indagini in tutto il mondo di questi agenti patogeni nella popolazione umana devono essere eseguite immediatamente, in modo che la conoscenza circa l'evoluzione e la diversità di questi agenti patogeni possono essere incorporati nei programmi di ricerca volti a migliorare le condizioni che causano.
 I batteriofagi potrebbero trattare E. coli senza danneggiare l'intestino,
I batteriofagi potrebbero trattare E. coli senza danneggiare l'intestino,
 Nuovo modello per il trapianto di microbioma vaginale
Nuovo modello per il trapianto di microbioma vaginale
 La malattia di Parkinson potrebbe essere prevenuta dai microbi intestinali
La malattia di Parkinson potrebbe essere prevenuta dai microbi intestinali
 In che modo gli sforzi di screening di massa hanno aiutato a identificare più casi di celiachia nei bambini
In che modo gli sforzi di screening di massa hanno aiutato a identificare più casi di celiachia nei bambini
 Non temere la colonscopia
Non temere la colonscopia
 Le risposte alimentari sono governate dal microbioma intestinale,
Le risposte alimentari sono governate dal microbioma intestinale,
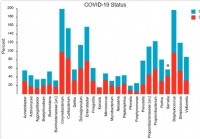 La composizione e la struttura del microbioma nasofaringeo si riferiscono alla gravità della malattia COVID-19
Le infezioni virali sono associate a cambiamenti nel microbioma del tratto respiratorio superiore/rinofaringeo (NP). Inoltre, molti studi ipotizzano la possibilità di super infezioni dovute allimmunit
La composizione e la struttura del microbioma nasofaringeo si riferiscono alla gravità della malattia COVID-19
Le infezioni virali sono associate a cambiamenti nel microbioma del tratto respiratorio superiore/rinofaringeo (NP). Inoltre, molti studi ipotizzano la possibilità di super infezioni dovute allimmunit
 E. coli superbatterio che si diffonde a causa della scarsa igiene del bagno,
non attraverso il cibo Un nuovo studio pubblicato su Le malattie infettive della lancetta il 22 ottobre, 2019, dice che un superbatterio comune che ne provoca più di 5, 000 casi di intossicazione al
E. coli superbatterio che si diffonde a causa della scarsa igiene del bagno,
non attraverso il cibo Un nuovo studio pubblicato su Le malattie infettive della lancetta il 22 ottobre, 2019, dice che un superbatterio comune che ne provoca più di 5, 000 casi di intossicazione al
 L'uso a lungo termine di antibiotici nei prematuri promuove batteri intestinali resistenti ai farmaci
I neonati molto prematuri sono spesso malati e richiedono un trattamento antibiotico per salvare le loro vite. Però, quando questa forma di terapia dura per 20 mesi o più, può influenzare il microbiom
L'uso a lungo termine di antibiotici nei prematuri promuove batteri intestinali resistenti ai farmaci
I neonati molto prematuri sono spesso malati e richiedono un trattamento antibiotico per salvare le loro vite. Però, quando questa forma di terapia dura per 20 mesi o più, può influenzare il microbiom