NMDA sowie andere Nitrosamin-Verunreinigungen werden seit einiger Zeit von der FDA untersucht, insbesondere in Blutdruckmedikamenten, den sogenannten Angiotensin-II-Rezeptorblockern. Hohe Nitrosaminkonzentrationen haben zum Rückruf mehrerer Marken dieser Medikamente geführt. Derzeit sind die in Ranitidin-Marken nachgewiesenen Nitrosaminkonzentrationen niedrig und die FDA würde Aussagen zum Rückruf veröffentlichen. Dies war ein Warnbericht.
Der Bericht sagt, „Patienten sollten darauf vertrauen können, dass ihre Medikamente so sicher wie möglich sind und dass der Nutzen der Einnahme jedes Risiko für ihre Gesundheit überwiegt. Obwohl NDMA in großen Mengen Schaden anrichten kann, Die von der FDA in vorläufigen Tests festgestellten Werte für Ranitidin übersteigen kaum die Mengen, die Sie in üblichen Lebensmitteln erwarten würden.“
Ranitidin ist ein weit verbreitetes rezeptfreies und verschreibungspflichtiges Medikament, das zur Klasse der H2-Blocker (Histamin-2) gehört und zur Verringerung der Säuresekretion im Magen nützlich ist. Es wird zur Behandlung von Sodbrennen verwendet, Verschlucken von Säure usw. Verschreibungspflichtiges Ranitidin wird zur Behandlung von Magen-Darm-Erkrankungen verwendet, Vorbeugung von Magen- und Zwölffingerdarmgeschwüren sowie Behandlung und Behandlung von GERD (gastroösophageale Refluxkrankheit).
Derzeit untersucht die FDA die Quelle von NDMA bei der Herstellung von Ranitidin und hofft, dass, wenn es entfernt wird, das Medikament könnte wieder für sicher erklärt werden. Der Bericht sagt, „Die FDA wird basierend auf den Ergebnissen der laufenden Untersuchung geeignete Maßnahmen ergreifen. Die Agentur wird weitere Informationen bereitstellen, sobald sie verfügbar sind.“
Die Warnung wiederholt, dass Personen die Einnahme von Ranitidin aufgrund dieser Warnung nicht abbrechen sollen. Personen, die ihre Ranitidin-Verschreibungen absetzen möchten, müssen mit ihrem Arzt über alternative Behandlungsmöglichkeiten sprechen. sagt die FDA.
Die FDA fügte in der Warnung hinzu, „Verbraucher und Angehörige der Gesundheitsberufe sollten alle Nebenwirkungen von Ranitidin dem MedWatch-Programm der FDA melden, um der Behörde zu helfen, den Umfang des Problems besser zu verstehen.“ Sie fordern die Verbraucher auf, einen Bericht online unter www.fda.gov/medwatch/report.htm auszufüllen und einzureichen und das entsprechende Formular auszufüllen und per Fax an 1-800-FDA-0178 einzureichen.
Am 24. September, die FDA gab erneut eine Warnung heraus, die Angehörige der Gesundheitsberufe und Patienten darauf aufmerksam machte, dass sie 14 Chargen verschreibungspflichtiger Ranitidin-Kapseln zurückgerufen hatten, die von Sandoz Inc. vertrieben wurden. Dieser Rückruf war auf die in den Chargen gefundenen Mengen an NDMA zurückzuführen.
Der amtierende FDA-Kommissar Ned Sharpless, M. D., in einer Erklärung heißt es, „Die FDA setzt sich dafür ein, dass die Medikamente, die die Amerikaner einnehmen, sicher und wirksam sind. Wir haben sofort mit dem Testen von Ranitidin-Produkten begonnen, nachdem wir von der potenziellen Verunreinigung erfahren hatten. Wenn wir Qualitätsmängel bei Arzneimitteln erkennen, die potenzielle Risiken für Patienten darstellen, Die FDA unternimmt alle Anstrengungen, um das Problem zu verstehen und der Öffentlichkeit so schnell und genau wie möglich unsere beste Empfehlung zu geben.“ „Wir werden weiterhin Untersuchungen durchführen und daran arbeiten, sicherzustellen, dass diese Arten von Verunreinigungen die akzeptablen Grenzen nicht überschreiten. damit die Patienten die benötigten Medikamente unbesorgt einnehmen können, " er fügte hinzu.
Im zweiten Bericht informierte die FDA Patienten und Angehörige der Gesundheitsberufe über Ranitidin, das von Sandoz hergestellt wurde, und sagte, dass, wenn der Patient eines der zurückgerufenen Medikamente einnimmt, er oder sie sollte die Rückrufanweisungen auf der FDA-Website befolgen. Der Bericht fügte hinzu, dass diejenigen Patienten, die keine nicht zurückgerufenen Marken von Ranitidin sind, dies weiterhin tun können. Der Bericht fügte hinzu, "Es ist wichtig, sich daran zu erinnern, dass nicht alles in den USA vermarktete Ranitidin zurückgerufen wird." In dem Bericht heißt es, dass Patienten, die OTC-Ranitidin einnehmen, andere Alternativen für ihre Symptome in Betracht ziehen können.
Janet Waldschnepfe, M. D., Direktor des Center for Drug Evaluation and Research der FDA, in einer Erklärung heißt es, „Wir setzen unsere Ermittlungen zusammen mit unseren internationalen Kollegen fort. und wir werden die amerikanische Öffentlichkeit über alle weiteren Rückrufe sowie die potenziellen Risiken der Einnahme von Ranitidin-Produkten informieren.“
Um die NDMA-Spiegel zu erkennen, hat die FDA ein Protokoll für die Aufsichtsbehörden und Hersteller veröffentlicht. Die NDMA-Spiegel in Ranitidin, das von diesen Herstellern hergestellt wird, müssen von ihnen anhand dieser Tests angegeben werden. Außerdem sollen sie die Proben zur Untersuchung durch die Wissenschaftler der Behörde an die FDA schicken.
 Kalorienbeschränkung führt zu Gewichtsverlust, kann aber pathogene Bakterien fördern
Kalorienbeschränkung führt zu Gewichtsverlust, kann aber pathogene Bakterien fördern
 SCD Lifestyle 2.0 ist da
SCD Lifestyle 2.0 ist da
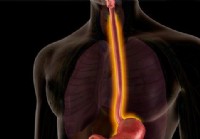 Ösophagitis (Schmerzen, Symptome, Ursachen, Grade und Heilung)
Ösophagitis (Schmerzen, Symptome, Ursachen, Grade und Heilung)
 Probiotika für gute Darmbakterien
Probiotika für gute Darmbakterien
 CED- und Morbus-Crohn-Ausschlussdiät (enerale Ernährung)
CED- und Morbus-Crohn-Ausschlussdiät (enerale Ernährung)
 7 Hormone, die jede Frau verstehen sollte
7 Hormone, die jede Frau verstehen sollte
 Was ist eine distale Gastrektomie?
Was ist eine distale Gastrektomie? Bei einer distalen Gastrektomie oder Antrektomie wird ein Teil des Magens entfernt und der Rest mit einer Öffnung in den Dünndarm genäht. Die Antrektomie (distal
Was ist eine distale Gastrektomie?
Was ist eine distale Gastrektomie? Bei einer distalen Gastrektomie oder Antrektomie wird ein Teil des Magens entfernt und der Rest mit einer Öffnung in den Dünndarm genäht. Die Antrektomie (distal
 Neun Mythen über Verdauungskrankheiten
Medizinische Gutachter und Herausgeber:Jay W. Marks, MD, und Melissa Conrad Stöppler, MD Forscher haben erst vor kurzem begonnen, die vielen, oft komplexen Krankheiten zu verstehen, die das Verdauung
Neun Mythen über Verdauungskrankheiten
Medizinische Gutachter und Herausgeber:Jay W. Marks, MD, und Melissa Conrad Stöppler, MD Forscher haben erst vor kurzem begonnen, die vielen, oft komplexen Krankheiten zu verstehen, die das Verdauung
 L-Glutamin:7 Gebote und Verbote für Menschen mit Leaky Gut &Autoimmunität
„Ewwww, bedeutet das, dass meine Kacke in meinen Körper ausläuft?“ Nur diejenigen unter Ihnen, die mit chronischen Darmproblemen zu tun haben, können solche Fragen zu schätzen wissen. Aber es ist ei
L-Glutamin:7 Gebote und Verbote für Menschen mit Leaky Gut &Autoimmunität
„Ewwww, bedeutet das, dass meine Kacke in meinen Körper ausläuft?“ Nur diejenigen unter Ihnen, die mit chronischen Darmproblemen zu tun haben, können solche Fragen zu schätzen wissen. Aber es ist ei