gástrico submucoso primária plasmocitoma nodular do estômago: uma variante pouco reconhecido de linfoma gástrico da arte abstracta da arte abstracta
plasmocitoma gástrica (GP) é uma variante rara de linfomas gástricos. Em caso excepcionais que um paciente apresenta GP, a lesão ocupa a camada da mucosa, na grande maioria dos casos. Aqui nós relatamos um caso de plasmocitoma nodular confinado à submucosa, sem evidência de pylori
infecção por Helicobacter (Hp). O paciente era um apresentador do sexo feminino de 59 anos de idade sem sintomas específicos. O tumor foi bem demarcada e consistia de uma proliferação monomórfica difusa de células plasmáticas com inúmeros folículos linfóides dispersas por todo o tumor. A superfície mucosa estava intacto e não associado com quaisquer nódulos tumorais. As células foram difusamente positivo para CD79a, Bob1, EMA e IgA e consistentemente negativo para CD3, CD19, CD20, PAX5, CD56, IgM e IgG. Além disso, em
hibridação in situ demonstrou clonalidade sob a forma de restrição da cadeia leve λ. Este padrão de proliferação nodular submucoso de plasmocitoma é pouco reconhecida e considerada como uma nova variante de linfoma
slides virtuais
O slide virtual (s) para este artigo pode ser encontrado aqui:. Http: //www. diagnosticpathol gia. diagnomx. /vs /3489998708673079
Palavras-chave
plasmocitoma solitário estômago fundo submucosa linfoma
linfomas extranodal zona marginal da mucosa tecido linfóide associado (linfoma MALT) e difuso de grandes células B UE linfomas estão bem documentados como os subtipos mais comuns de GI linfomas [1]. Dentro deste contexto, é também importante notar que o linfoma MALT está frequentemente associado a pylori
infecção por Helicobacter (Hp). Além de apresentar uma grande população de linfócitos que formam lesões linfoepiteliais distintas, cerca de um terço dos casos de linfoma MALT vai apresentar com diferentes graus de diferenciação plasmocítica [2]. doença intestinal pequena imunoproliferativa (IPSID) em particular, que é um subtipo de linfoma MALT e observado primariamente no Médio Oriente, tipicamente mostra marcante diferenciação plasmocitário [2].
Na extremidade oposta do espectro, plasmacitoma extraóssea (PE) , que é caracterizada por uma dilatação localizada e monoclonal de células plasmáticas em outros tecidos do que o osso, surgem no tracto respiratório superior, que inclui a orofaringe, nasofaringe, dos seios nasais e da laringe e [2]. O próximo local mais freqüente de ocorrência lesão de massa é o estômago; no entanto, isso é extremamente rara e representa menos de 5% de todos os EPs [3]. Além disso, a grande maioria dos plasmacitomas gástricas convencionais têm sido documentados principalmente na camada da mucosa [4-6].
Detalhes Este relatório de um caso raro de uma excepcionalmente plasmacitoma nodular ocupando a submucosa gástrica sem uma infecção subjacente Hp. Para o melhor de nosso conhecimento, apenas dois casos semelhantes foram relatados até o momento e, portanto, esta condição pode ser efetivamente definida como uma entidade pouco reconhecido de linfoma gástrico primário.
Apresentação do caso
história clínica
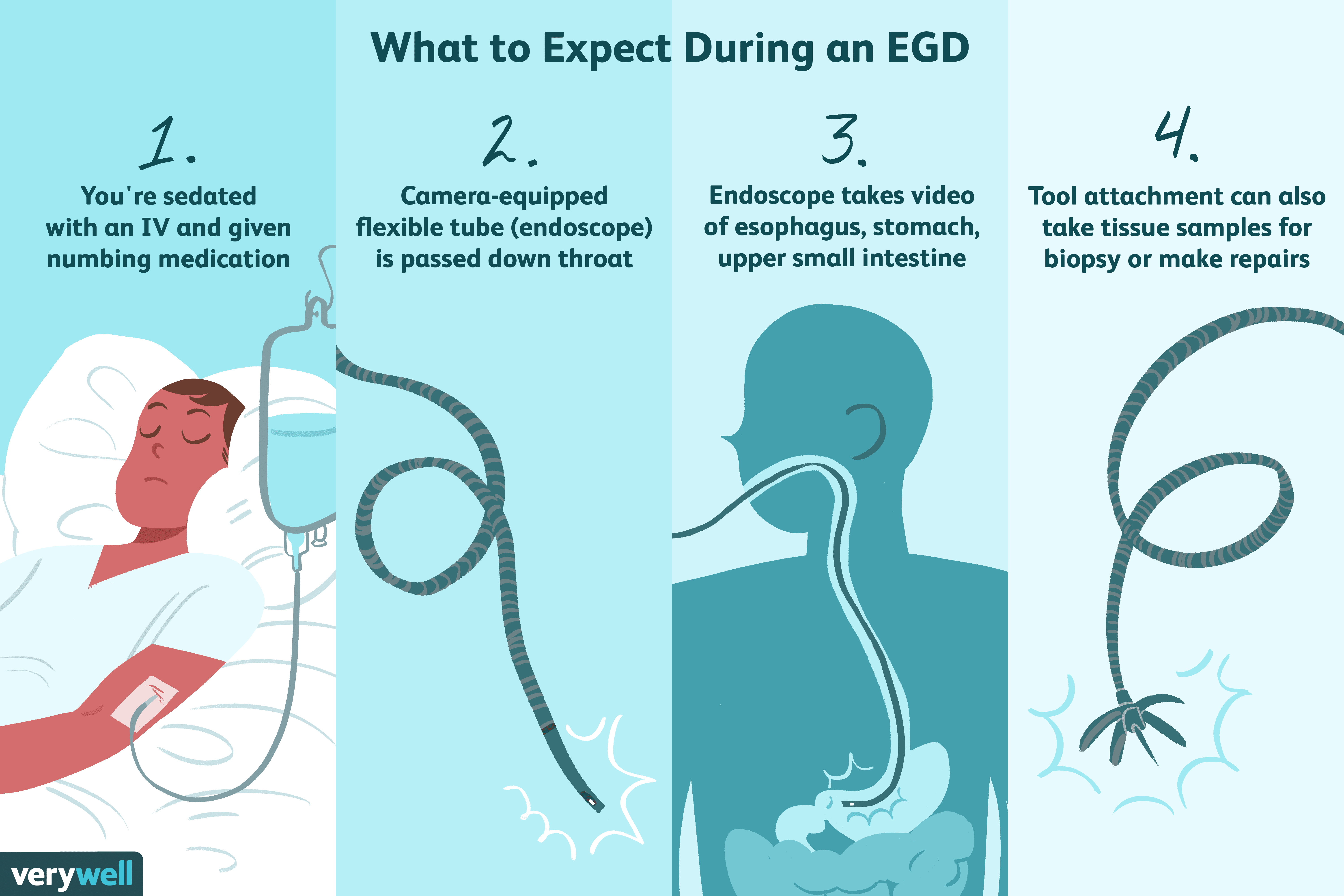
Um 59- fêmea com dois anos com um passado histórico médico do carcinoma papilar da tiróide foi encaminhada ao nosso hospital devido a um tumor gástrico identificados durante uma esophagogastroduodenoscopic triagem em massa (EGD) exame, que revelou uma lesão bem elevado, a 2 cm de diâmetro, ocupando as paredes posteriores do corpo gástrico (Figura 1). Nenhuma mudança ulcerativa foi observada na superfície da mucosa, o paciente estava livre de quaisquer sintomas que acompanham e resultados do estudo de laboratório estavam todos dentro dos limites normais. Com base em um diagnóstico de tumor na submucosa não especificado de outro modo, foi realizada uma gastrectomia parcial. A Figura 1 esofagogastroduodenoscopia. Bem demarcadas, lesões elevadas na parede posterior do corpo gástrico.
Características do mieloma múltiplo não eram aparentes e níveis séricos de cálcio, hemoglobina e creatinina do doente estavam dentro dos limites normais. Sem resultados significativos em um exame de raio-X do esqueleto foram anotadas e tomografia por emissão de pósitrons /tomografia computadorizada (PET /CT) não apresentaram outras lesões. Finalmente, soro e proteína na urina eletroforese produziu resultados normais. Nenhum tratamento adicional foi realizada. O presente estudo foi aprovado pelo Comitê de Ética da Universidade de Kobe Graduate School of Medicine.
Características brutas
A mucosa superfície do tumor era lisa e de aspecto normal. O tumor ressecado medido 2,0 × 1,5 × 1,5 cm foi bem demarcada, e consistiu de uma massa macia e elástica esbranquiçada localizado na camada submucosa. O tumor tinha uma superfície de corte homogéneo, de cor castanho e foi focally hemorrágico.
Microscópica apresenta
Histologicamente, o tumor foi relativamente bem demarcada e totalmente localizado na camada submucosa (Figura 2). A superfície mucosa que cobre o tumor estava intacto e não foi associada com a própria nódulo tumoral, que consistia de uma proliferação monomórfica difusa de células plasmáticas (Figura 3). Os núcleos das células tumorais foram perifericamente deslocadas e levemente pleomorfo. Poucas células tumorais continham corpos DUTCHER nos seus núcleos. Muitos folículos linfóides foram espalhados por todo o nódulo tumoral (Figura 4) e contou com zonas manto bem formados e centros foliculares que procuram normais, que consistiam principalmente de grandes centroblastos com dispersas macrófagos corpo tingible. Giemsa coloração não mostrou Hp aderir à superfície da mucosa gástrica. A biópsia espécime de medula óssea foi normal e não ofereceu nenhuma evidência sugestiva da proliferação de células plasmáticas. Os achados imunohistoquímicos são fornecidos na Figura 5. As células tumorais foram difusamente positivo para CD79a, Bob1, EMA, CD138, MUM-1 e IgA e negativo para CD5, IgM e IgG. Embora os folículos linfóides e um pequeno número de células B reactivas dispersos na zona interfolicular foram positivas para CD20, as células tumorais foram negativas para CD20, CD19 e CD56. A imunofenotipagem dos centros germinais dos folículos linfóides dispersas era normal: CD10 sentido bcl-2 de polaridade positiva, negativa e preservados em Ki67 (dados não mostrados). In situ
hibridação para κ e cadeias leves de imunoglobulina X revelou restrição da cadeia leve λ nas células tumorais. Além disso, a coloração não revelou D2-40 estruturas nos seios, como no nódulo de tumor, indicando que o nódulo difere a partir do nó de linfa. Figura 2 O tumor forma um nódulo gástrico submucoso (HE). O encarte mostra mucosa normal.
Figura 3 O tumor é constituído por proliferação difusa de células plasmáticas moderadamente pleomórficas. O encarte mostra corpos DUTCHER (HE).
Figura 4 folículos linfóides Numerosos ficava entre as células tumorais (HE).
Figura 5 estudos de imuno-histoquímica. a) CD79a é difusamente positivo. As células que demonstram forte positividade são células B reactivas. b) CD20 só é positivo em células B não neoplásicas nos folículos dispersos, e em pequenas quantidades em células reativas interfolicular. c) Bob-1, d) EMA, e) CD138 e f) MUM-1 são difusamente positivo. As células tumorais são positivos para g) IgA, e negativo para h) IgG e i) IgM. j) D2-40 coloração não revelaram estruturas nos seios, como no nódulo tumoral. k) compósitos de células tumorais descreve ausência de expressão da cadeia leve κ, e l) difundir expressão da cadeia leve citoplasmática λ.
Dados os resultados de laboratório listados acima, as lesões foram diagnosticados como submucosa primária plasmacitoma nodular do estômago.
Discussão
neste relatório nós descrevemos um caso extremamente raro de um plasmocitoma nodular principal desenvolver na submucosa gástrica, o que provavelmente constitui uma entidade pouco reconhecido de linfoma gástrico primário.
no que diz respeito à proliferação neoplásica de células plasmáticas no estômago , plasmocitoma primário deve sempre ser distinguidos de outras doenças linfoproliferativas com diferenciação plasmocítica, tais como mieloma das células plasmáticas, linfoma linfoplasmocitário (LPL), variante de células plasmáticas doença de Castleman (PC-CD), e linfoma MALT.
O sistêmica mandados de exame ósseo aspiração ou biópsia da medula, a fim de distinguir esta condição de envolvimento gástrica de mieloma das células plasmáticas óssea. No presente caso, não há sintomas ou achados sugestivos de mieloma das células plasmáticas foram documentados. Em termos de imuno-histoquímica, 67-79% dos casos ósseas mieloma das células plasmáticas demonstrar CD56 positivo [7]. Embora os resultados são, em última análise inconclusivos devido ao número amostra limitada, CD56 negatividade talvez pudesse ser explorado como uma ferramenta para diferenciar esta doença de mieloma das células plasmáticas.
O estômago é um local relativamente comum para os linfomas extranodais. Enquanto os linfomas de células T e de células T /NK também são conhecidas por afectarem de forma esporádica do estômago, a maioria dos linfomas gástricos primários são da linhagem de células B [1, 8]. linfomas MALT mais frequentemente surgem no tracto GI e morfologicamente heterogéneas de células consistem pequenas B, células monocitóides, pequenos linfócitos, assim como imunoblastos dispersos e células centroblast semelhante [9]. Normalmente, células de linfoma MALT neoplásicas expressam marcadores de linhagem de células B, e são negativos para a ciclina D1, CD23, CD10 e CD5, com raras exceções [10]. diferenciação plasmocítica é frequentemente presentes em linfomas MALT gástricos; no entanto, eles desenvolvem na mucosa e estão intimamente associados com o envolvimento do epitélio glandular. Assim, eles são frequentemente referidos como lesões linfoepiteliais. Além disso, a maioria dos casos (62-77%) está associada com a infecção pelo Hp [9]. O caso apresentado, no entanto, revelaram um padrão completamente distinta da doença:. Bem demarcadas nódulos "submucosos", uma população puramente plasmocítica de células tumorais, e nenhuma infecção subjacente Hp
PC-CD geralmente é sintomático, com febre, suores nocturnos, perda de peso e lesões múltiplas. PC-CD do trato gastrointestinal é extremamente raro e houve apenas um relato de caso documentando uma lesão polipóide 4 cm de diâmetro envolvendo o reto [11]. Normalmente, PC-CD mostra a proliferação acentuada de vasos, além de infiltração plasmocitária de destaque nas áreas interfolicular. Os folículos linfóides apresentam um padrão reativo com características hialina vasculares raras e embarcações penetraram. Além disso, o PC-CD geralmente apresenta-se com uma grande variedade de células B e células de plasma são geralmente policlonal; no entanto, tem havido relatos que detalham evidências imunofenotípica de Ig restrição cadeia pesada e leve, bem [12]. Portanto, um exame morfológico metódica dos folículos e áreas interfolicular é imperativo distinguir PC-CD de plasmocitoma. No presente caso, nenhuma das características distintivas associadas a foram identificados PC-CD.
LPL geralmente envolve a medula óssea e a maioria dos pacientes possuem uma paraprotein IgM no soro. Histologicamente, há uma proliferação relativamente monótona de pequenos linfócitos, células plasmáticas e linfócitos plasmocitoides com relativamente poucas células transformadas. corpos DUTCHER, o aumento dos mastócitos e hemossiderina são também características típicas, e a cadeia leve κ é expresso com mais freqüência do que a cadeia leve λ. Há um caso relatado de LPL que ocorre no estômago; no entanto, esta acabou por ser descartada como uma lesão gástrica de envolvimento secundário [13]. No presente caso, o paciente apresentava nenhum sintoma, e não apresentava sinais de linfadenopatia ou soro paraproteínas. Além disso, os núcleos das células do tumor foram mais pleomorfo e as células linfóides não plasmacytic faltava um componente neoplásica.
Normalmente linfomas MALT ou LPLs express IgM, enquanto plasmacitomas expressam IgG. No caso aqui relatado, as células tumorais foram difusamente positivos para IgA. Embora plasmocitoma expressar IgA é um fenômeno raro, Shao et al relatou recentemente a ocorrência de um plasmocitoma expressar IgA, que contou com idade jovem na apresentação, o baixo risco de progressão para mieloma das células plasmáticas, e associação com doença auto-imune ou disfunção imune [14]. Em contraste, no nosso caso, a paciente não apresentava nenhuma das características clínicas descritas por Shao et al.
Muitos casos de plasmocitoma gastric relatado anteriormente eram doenças essencialmente mucosa [4-6] enquanto nosso caso mostrou uma completamente diferente padrão de proliferação. Consideramos, portanto, que seja uma variante pouco reconhecido e rara de plasmocitoma gástrico. Até à data, apenas dois casos foram relatados plasmocitomas gástricas localizados inteiramente na submucosa como descrevemos no presente caso [15, 16]. Uma comparação entre os casos e o nosso é mostrado na Tabela 1. A média de idade dos três casos, que consiste em dois homens e uma mulher, foi de 61 anos (intervalo: 57-67 anos). O tamanho médio do tumor foi de 2,1 cm (intervalo: 0,7-4,1 cm). Ressecções foram realizados e nenhum dos casos mostrou qualquer evidência de recorrência do tumor ou o desenvolvimento de mieloma sistêmica após a primeira treatment.Although é necessário um corpo maior de casos para chegar a quaisquer conclusões definitivas, a experiência com estes três casos sugere que a terapia agressiva não é necessário para pacientes que tenham sido submetidos a excisão cirúrgica completa. linfomas MALT, juntamente com certos casos de plasmocitomas gástricas associadas com a infecção pelo Hp foram supostamente curada com Hp erradicação [17, 18]. No entanto, não foi detectada a infecção pelo Hp no nosso caso, ou qualquer um dos outros casos relatados anteriormente. Portanto, a erradicação do Hp não é, presumivelmente, um meio eficaz de tratamento para este subtype.Table 1 Resumo da submucosa gástrica plasmocitoma
Idade
Sex
Região no estômago
Tamanho (cm)
Terapia
sobrevivência livre de doença (ano)
Roost. et al (2007)
67
masculino
Ângulo
0,7
EMR *
2,5
Navin. et al (2010)
57
masculino
curvatura maior
4.1
gastrectomia
nenhuma informação
Nosso caso
59
feminina
Corpus
1,5
ressecção parcial
1,5
* EMR, a ressecção endoscópica da mucosa.
dispersados folículos linfóides "reativo" no nódulo tumoral são a definição de achados característicos no caso em apreço. Embora os dois casos da submucosa plasmacitoma anteriormente referidos foram também bem circunscrito, o tumor descrito neste estudo apresentado com uma consistência pura de células do plasma, sem a formação de folículos linfóides. Embora a razão exata a respeito de porque este tumor apresentados com folículos linfóides scattererd não é clara, uma infecção subjacente ou envolvimento ganglionar estão algumas explicações plausíveis. Sabe-se que alguns agentes infecciosos, incluindo HP ou vírus Epstein-Barr induzir mudanças inflamatória com folículos linfóides no estômago [19, 20]; No entanto, Hp e hibridização in-situ para o vírus de Epstein-Bar de ARN codificada por vírus (EBER) foram ambos negativos neste caso. Além disso, D2-40 imunocoloração não revelou quaisquer estruturas SINUS como bem desenvolvidas dentro do nódulo de tumor. Deve também ser notado que os nódulos linfáticos não deve estar presente na submucosa normal. A partir destes resultados, pode-se deduzir que a possibilidade de envolvimento ganglionar é improvável. No entanto, a possibilidade de envolvimento ganglionar não pode ser excluída devido à presença de estruturas periféricas finas semelhantes a cápsulas, a morfologia bem demarcadas, e alguns vasos D2-40-positiva dentro do nódulo. Além disso acúmulo de casos semelhantes e suas análises subsequentes são necessários para esclarecer a extensão destas morfologias únicas.
Conclusões
Para resumir, este relatório descreve um caso extremamente raro de um plasmocitoma solitário primária situados exclusivamente na submucosa gástrica. Este padrão de proliferação é pouco reconhecida e muito provavelmente constitui uma nova variante de linfoma gástrico. Embora o prognóstico parece ser excelente, um maior número de casos semelhantes é necessária a fim de esclarecer as características clinicopatológicas desta variante.
Consentimento
consentimento informado foi obtido de paciente para a publicação deste relato de caso e as imagens que acompanham . Uma cópia da autorização escrita está disponível para análise pelo Editor-in-Chief da patologia de diagnóstico.
Declarações
Agradecimentos
Os autores gostariam de agradecer ao Dr. Richard H. Kaszynski no Departamento de Medicina Legal, Kobe University Graduate School of Medicine, em sua edição e contribuição construtiva na formulação deste manuscrito.
Autores 'arquivos enviados originais para imagens
Abaixo estão os links para os autores' arquivos enviados originais de imagens. 'arquivo original para a figura 1 13000_2012_709_MOESM2_ESM.tiff Autores' 13000_2012_709_MOESM1_ESM.tiff Autores arquivo original para 'arquivo original para a figura 3 13000_2012_709_MOESM4_ESM.tiff Autores' figura 2 13000_2012_709_MOESM3_ESM.tiff Autores arquivo original para a figura 4 arquivo original 13000_2012_709_MOESM5_ESM.tiff Autores 'para a figura 5 'arquivo original para a figura 6 13000_2012_709_MOESM7_ESM.jpeg Autores' 13000_2012_709_MOESM6_ESM.jpeg Autores arquivo original para a figura 7 interesses concorrentes
não temos quaisquer interesses concorrentes para o nosso manuscrito. contribuição
dos autores
MK foi responsável por dados coleta e elaborou o manuscrito. TI ajudou a revisar este manuscrito. CH, YK, FK, SH ajudou ao diagnóstico. HM era médico assistente do paciente e fez contribuição para aquisição da dados clínicos. Todos os autores leram e aprovaram o manuscrito final.
 Barras de proteína macia
Um dos problemas mais comuns que vemos quando as pessoas começam SCD, GAPS ou Paleo é que elas tentam recriar os alimentos que costumavam comer da Dieta Americana Padrão. Inevitavelmente, as receitas
Barras de proteína macia
Um dos problemas mais comuns que vemos quando as pessoas começam SCD, GAPS ou Paleo é que elas tentam recriar os alimentos que costumavam comer da Dieta Americana Padrão. Inevitavelmente, as receitas
 Consultando um gastroenterologista para diarreia crônica
Todo mundo tem um ataque ocasional de diarréia. Pode ser estresse ou algo que você comeu; no entanto, e se a sua diarreia não desaparecer? Se você está com diarreia consistentemente por várias semanas
Consultando um gastroenterologista para diarreia crônica
Todo mundo tem um ataque ocasional de diarréia. Pode ser estresse ou algo que você comeu; no entanto, e se a sua diarreia não desaparecer? Se você está com diarreia consistentemente por várias semanas
 Como saber se tenho cálculos biliares?
O que são cálculos biliares? Os cálculos biliares são depósitos pequenos e duros que se formam na vesícula biliar a partir da bile que se cristaliza. Dor abdominal, náusea, vômito, febre, calafrios
Como saber se tenho cálculos biliares?
O que são cálculos biliares? Os cálculos biliares são depósitos pequenos e duros que se formam na vesícula biliar a partir da bile que se cristaliza. Dor abdominal, náusea, vômito, febre, calafrios