die funktionellen Zusammenspiel von Helicobacter pylori
Faktoren mit Magen-Epithelzellen induziert einen mehrstufigen Prozess in Pathogenese
Zusammenfassung
Infektionen mit der menschliche Erreger Helicobacter pylori
(H
. pylori
) kann zu schweren Magenerkrankungen führen von chronischer Gastritis und Geschwüren zu neoplastischen Veränderungen im Magen reichen. Entwicklung und den Fortschritt der H
. pylori
assoziierte Erkrankungen sind durch vielfältige Bakterien Faktoren bestimmt. Viele von ihnen interagieren direkt mit Wirtszellen oder spezifische Rezeptoren erfordern, während andere die Host-Zytoplasma eingeben zelluläre Funktionen zu entgleisen. Mehrere Adhäsine (z BabA, SabA, AlpA /B, oder OIPA) engen Kontakt mit dem Magen-Epithel als wichtigen ersten Schritt in persistent Besiedlung etablieren. Lösliche H
. pylori
Faktoren (zum Beispiel Urease, VacA oder HtrA-) vorgeschlagen worden das Überleben der Zellen und interzellulären Adhäsionen zu verändern. Über eine Typ-IV-Sekretionssystem (T4SS), H
. pylori
transloziert auch die Effektor Cytotoxin-assoziierte Gen A (CagA) und peptidoglycan direkt in die Wirts Zytoplasma, wo Krebs- und entzündungsbedingte Wege Signaltransduktion dereguliert werden kann. Durch diese vielfältigen Möglichkeiten der Interaktion mit Wirtszellen, H
. pylori
stört die komplexen Signaltransduktionsnetzwerken in seinem Wirt und vermittelt ein mehrstufiges Pathogenese.
Bewertung
Die Interaktion zwischen Erregern und Gewebe- oder organspezifische Zielzellen in ihrem Wirt die Gründung und Entwicklung bestimmt von Infektionskrankheiten. Daher müssen Krankheitserreger angepasst belichten, sondern spezialisierte Faktoren, die die Abwehrmechanismen des Wirts an der Gewebeoberfläche zu überwinden. Im Verdauungstrakt ist die Magenschleimhaut durch einen dicken Schleimschicht zum Schutz der Epithel von Protein-lysierenden Enzymen, Magensäure bedeckt und schließlich Speisebrei, die auch unerwünschte Bakterien und Krankheitserregern enthalten kann. Die Ausbildung dieser erste wirksame Barriere, Epithelzellen zeigen eine apikal-basalen Organisation, die von tight junctions in erster Linie beibehalten wird, die Einhaltung Kreuzungen und einem streng geregelten Aktin-Zytoskelett [1, 2]. Funktionelle tight junctions sind entscheidend für die Aufrechterhaltung der epithelialen Polarität und Zell-Zell-Adhäsion und eine parazellulären Barriere bilden, die den freien Durchgang der Moleküle ausschließt. Tight Junctions sind aus mehreren Arten von Transmembranproteinen (zB Occludin, Claudinen, junctional Adhäsionsmoleküle [JAMs]), die Bindung an zytoplasmatische peripherer Proteine (zB Zonula occludens [ZO] protein-1, -2 und -3, Cingulin oder multi- PDZ-Protein-1 [MUPP1]) und verbinden die Transmembranproteine an das Aktin-Zytoskelett. Adherence junctions vermitteln interzellularen Adhäsionen zwischen benachbarten Zellen, steuern die Aktin-Zytoskelett und deshalb weisen anti-Tumor-Eigenschaften. Sie bestehen aus dem Transmembranprotein E-Cadherin, die benachbarte Epithelzellen mit der intrazellulären Actin-Cytoskelett überbrückt. Dies beinhaltet eine Signalkomplex, bestehend aus β-Catenin, p120-Catenin, α-Catenin und epithelialen Protein verloren in Neoplasma (EPLIN), die an die intrazelluläre Domäne von E-Cadherin rekrutiert wird. Diese dynamischen interzellulären Verbindungen sind von entscheidender Bedeutung für die Integrität des Magen-Epithel und zum Schutz gegen eindringende Krankheitserreger [1, 2]. Helicobacter pylori
(H
. Pylori
) ist eine bakterielle Klasse-I Karzinogen [3], die das Magenepithel von Menschen als einzigartige Nische spezifisch kolonisiert, wo sie Entzündungsstörungen induzieren kann (zB Geschwüren, chronische Gastritis, etc.) und maligne neoplastische Erkrankungen (Malt [MALT] Lymphom und Magenkrebs) [4, 5]. Um die feindlichen Umgebung im Magen widerstehen, H
. pylori
hat hoch entwickelte Mechanismen entwickelt, um ein Leben lang Infektionen im Magen festzustellen, ob nicht therapeutisch ausgerottet. Deshalb ist es als eine der erfolgreichsten bakterielle Pathogene angesehen wird. H
. pylori
induziert Gastritis in allen infizierten Patienten, aber nur eine Minderheit von etwa 10-15% leidet an klinischen Symptomen. Der Grund für die unterschiedlichen Reaktionen auf H
. pylori
ist nicht eindeutig geklärt, aber viele Berichte weisen auf individuellen genetischen Empfindlichkeiten des Wirtes zu H
. pylori
assoziierte Erkrankungen. Dementsprechend genetischen Polymorphismen mit einem erhöhten Risiko für Magenkrebs assoziiert sind in Gene kodierend Interleukine (z.B. IL-1β), Tumornekrosefaktor (TNF), Cyclooxygenase-2 (COX2) und andere Wirtsfaktoren [6, 7] identifiziert. Abgesehen von Wirtsfaktoren,
H. pylori
Isolate beherbergen verschiedene Muster von genetischen Elementen für bakterielle Faktoren kodieren, die in einem dauerhaften Besiedlung und Pathogenese entscheidend beteiligt sind. Einige von ihnen haben bereits als Virulenzfaktoren definiert worden [8], während andere als wichtige Nische und Kolonisierung Determinanten dienen [9] oder werden noch untersucht für ihre pathologische Relevanz.
In den letzten drei Jahrzehnten wurden bemerkenswerte Fortschritte im Verständnis der Pathogenität bezogenen Faktoren von H
hergestellt. pylori
und ihre funktionelle Interaktion mit Magen-epithelialen Zell-Komponenten. Diese Virulenz-Faktoren werden entweder sekretiert, membranassoziierte oder in das Cytosol von Wirtszellen transloziert, wo sie direkt mit den Wirtszellfunktionen (Abbildung 1) stören. Als Folge ihrer unterschiedlichen Stellen während des Infektionsprozesses,
H. pylori
ist in der Lage eine Vielzahl von Mechanismen zu nutzen Host zellulären Prozesse zu manipulieren und Signalkaskaden zu deregulieren. Der Einfluss von H
. pylori
dieser Signalwege führt zu Adhärenz, Induktion von proinflammatorischen Reaktionen durch Zytokin /Chemokin-Freisetzung, die Apoptose, Proliferation und eine ausgeprägte motogene Antwort als charakterisiert in vitro
. Zusammengenommen führen diese schließlich in einem dauerhaften Besiedlung, schwere Entzündungen, Störungen der epithelialen Barrierefunktion und möglicherweise Magenkrebs (Abbildung 1). Diese Effekte stammen aus selektiven Erreger-Wirt-Wechselwirkungen, die in diesem Bericht zusammengefasst wurden, um eine umfassende Übersicht über die große Anzahl von spezialisierten bakteriellen Faktoren und wie H
zu geben. pylori
nutzt sie die Magen-Epithel zu manipulieren. Viele dieser Faktoren wirken kooperativ, was schließlich zu einem komplexen Szenario der Pathogenese bezogenen Signalereignisse führen. Abbildung 1 zellulären Reaktionen auf H. pylori-Kolonisierung auf einer polarisierten Epithel. H
. pylori
drückt membrangebundene Faktoren, sondert Faktoren und nutzt eine Typ-IV-Sekretionssystem (T4SS) zu Effektoren injizieren. Diese tragen zur Adhäsion oder Signaltransduktionswege induzieren zur Induktion von proinflammatorischen Zytokin-Freisetzung führt, Apoptose, Zellbeweglichkeit oder Proliferation. . Dieses Netzwerk verschiedener Signalwege und zellulären Reaktionen werden bei der Etablierung einer persistierenden Infektion, Entzündung und Zerstörung der epithelialen Polarität und Integrität einen Beitrag zur Entwicklung von Gastritis, Geschwüre und Magen-malignen Erkrankungen
Membran-assoziierten Faktoren beteiligt: Adhäsine und darüber hinaus
Trotz Peristaltik des Magens und den Transport von Speisebrei, H
. pylori
stellt eine starke Interaktion mit Epithelzellen. In der Tat, die Haftung von H
. pylori
gilt als der erste wichtige Schritt in der Pathogenese im Magen zu sein. Die große Gruppe der äußeren Membranproteine (OMP) enthält einige Adhäsine (zB Blutgruppen-Antigen-bindenden Adhäsin [BabA], Sialinsäure Adhäsin [SabA], die Einhaltung-assoziierten Lipoprotein A und B [AlpA /B] und Außen inflammatorische Protein A [OIPA]), die von H
Mediate Bindung. pylori
auf der Wirtszellmembran und andere Faktoren (beispielsweise Lipopolysaccharid [LPS] und Flagellin), die Entzündungsreaktionen in Wirtsgewebe (2a) auslösen können. Abbildung 2 Modell von H. pylori-Faktoren mit Wirtszellen interagieren. (A) Auf der apikalen Seite des polarisierten Epithel H
. pylori
gründet die erste Einhaltung. SabA, BabA, AlpA /B, OIPA, HopZ, Horb, usw. werden als wichtige Adhäsine betrachtet, die Zell-Rezeptoren binden Host (z Leb, sLex, Laminin) und könnte NF-кB oder MAPK Aktivität beitragen. (B) H
. pylori
sondert VacA, die Poren in den Host-Membranen und lokalisiert auf Mitochondrien bildet, wo sie mit Apoptose-bezogenen Prozesse stören können. Darüber hinaus die zelluläre Barrierefunktion VacA könnte durch Tight Junctions zu beeinflussen beeinflussen; ein Effekt, der auch für löslichen Urease vorgeschlagen worden. Zusammen mit H
. pylori
-secreted HtrA-, die direkt spaltet die Einhaltung Kreuzung Molekül E-Cadherin, H
. pylori
stört effizient die epithelialen Barriere. Der T4SS spritzt den bakteriellen Faktor CagA. Auf der apikalen Seite der polarisierten Zellen, könnte CagA über Phosphatidylserin und Cholesterin translozieren. Im Cytosol von H
. pylori
infiziertem Zellen zeigt CagA hemmende Wirkung auf VacA-vermittelte Apoptose und die Integrität der engen und Einhaltung Junctions. HtrA-getriggerten E-cadherin-Spaltung kann durch H
verbessert werden. pylori
-induzierte MMPs und konnte die Destabilisierung der Einhaltung komplexer intrazellulärer β-Catenin und p120-Catenin zusammengesetzt erhöhen. Disruption des Komplexes E-Cadherin könnte an Tumor-assoziierte Zielgenexpression in den Zellkern und /oder zur Regulation des Actin-Zytoskeletts während der Zell morphologischen Veränderungen und Motilität beitragen. (C) Integrine sind an der basolateralen Seite eines polarisiertes Epithel exprimiert und könnte durch die T4SS Adhäsin CAGL nach Aufschluß der interzellularen Adhäsionen kontaktiert werden. CagA transloziert über α5β1-Integrinen und wird Tyrosin schnell phosphoryliert. Phosphoryliert CagA dereguliert dann Wege Signaltransduktion, Änderungen in der Genexpression führt, und interferiert stark mit dem Zytoskelett-Umlagerung, die für die motogene Reaktion auf H
wichtig ist. pylori
. Peptidoglycan gilt als ein anderer Effektorzellen zu sein, die Nod1 bindet, wodurch die Aktivierung der NF-кB Signalwege.
Obwohl Bakterienadhärenz für H
von entscheidender Bedeutung ist. pylori
Pathogenese, Daten über die direkten Auswirkungen der oben genannten Adhärenzfaktoren zeigt Signalwege sind rar. Dies zeigt, daß canonical Adhäsine nicht direkt Signalgebung zu aktivieren, sondern eine enge Wechselwirkung zwischen H
vermitteln. pylori
und der Host-Zielzelle, die wahrscheinlich den Weg für zusätzliche bakterielle Faktoren Pflasterung mit ihren verwandten Rezeptoren zu interagieren. Zusätzlich zu OMP und Adhäsine, Flagellin und LPS weit wurden untersucht ihre Rolle in der H
zu adressieren. pylori
Pathogenese. Im allgemeinen sind Flagellin und LPS wichtige Faktoren in vielen anderen bakteriellen Infektionen, aber es ist unklar, inwieweit die beiden Faktoren, die zu H
beitragen. pylori
-induzierten Ereignisse signalisieren. Im Gegensatz zu dem Flagellin von anderen bakteriellen Pathogenen,
H. pylori
Flagellin hat nur eine sehr geringe Kapazität Toll-like-Rezeptor 5 (TLR5) -abhängigen Interleukin-8 (IL-8) release [10] zu stimulieren. Dies wurde durch die Erkenntnis bestätigt, daß H
gereinigt. pylori
Flagellin ist ein schlechter Ligand für TLR5 [11]. Nur wenige Informationen über die Auswirkungen von H
zur Verfügung. pylori
LPS auf den Epithelzellen, eine noch nicht definierte Rolle in der H
angibt. pylori
auch infiziertem Epithel. Jedoch wurde gefunden, daß H
vorgeschlagen. pylori
LPS könnte eine TLR2-Agonist in Magen MKN45-Zellen sein, einen Beitrag zur Aktivierung des nukleären Faktors kappa B (NF-кB) und Chemokin-Expression unabhängig von dem kanonischen LPS-Rezeptor TLR4 [12]. Es wurden jedoch mehrere Faktoren auch H
etabliert. pylori
Adhäsine, die das Potenzial haben, Signaltransduktionswege zu verändern, entweder durch Bindung direkt an Zelloberflächenrezeptoren oder wirken indirekt, bringen andere bakterielle Faktoren in der Lage, mit Zelloberflächenstrukturen zu interagieren, die normalerweise die Kapazität für die Signalübertragung fehlt.
Blutgruppen-Antigen-bindenden Adhäsin (BabA)
H
. pylori
Adhäsion wurde mit der Anwesenheit von fucosylierten Blutgruppenantigene [13] und dem OMP BabA korreliert wurde anschließend als erste Adhäsin von H
identifiziert. pylori
, die auf dem Epithel [14] zu den fukosylierte Blutgruppe 0-Antigene Lewis B (Le
b) und die damit verbundene H1 bindet. Allerdings wird die Bindungsspezifität von BabA zu Blutgruppe 0-Antigene auf bestimmte H
beschränkt. pylori
Stämme, bezeichnet als "Spezialist" Stämme, während BabA von "Generalisten" Stämme gleichermaßen bindet fukosylierte Blutgruppe A-Antigene [15]. Vor kurzem wurde Globo H hexaglycosylceramide als zusätzliche BabA Bindungspartner vorgeschlagen, die eine Rolle bei der Infektion von nicht-secretor Individuen [16] spielen könnten. Interessanterweise wurden Fachstämme überwiegend in den südamerikanischen Ländern, wo die Blutgruppe 0 Phänotyp in der lokalen Bevölkerung vorherrscht. Diese Anpassungsfähigkeit in der Bindungsspezifität von BabA könnte zum Verlust der selektiven Druck auf die Blutgruppe A und B Bindung, anstatt aktive Auswahl von Fach Stämme zurückzuführen, für Affinitäten in Fach Stämme Bindung nicht mit denen von Generalisten Stämme auszeichnen [15]. Die Analyse der genetischen Grundlage der BabA ergab zwei BabA loci (BabA1 und BabA2, von denen BabA1 nicht exprimiert [17]) und ein eng verwandtes paraloge Babb locus [14]. Es wurde vorgeschlagen, dass BabA Expression über Phasenvariation und Rekombination mit dem Babb Locus reguliert wird, wie mehrere Studien Verlust- und gain-of-function Mutationen in vitro und in vivo
[14, gezeigt haben, 18- 20]. Darüber hinaus hat die genetische Konfiguration der bab
Gene wurden mit Vorzugs Lokalisation im Magen und der BabA /B Einstellung korreliert mit dem höchsten Risiko für Magenkrebs [21].
BabA vermittelte Adhäsion von H korreliert
. pylori
zu Magenepithelzellen könnte CagA-Translokation und die Induktion der Entzündung [22] zu verbessern. Darüber hinaus Triple-positive klinische H
. pylori
Isolate (BabA +, VacAs1 +, CagA +) zeigen größere Kolonisationsdichten, erhöhte Werte von Magen-Entzündung und eine höhere Inzidenz von intestinale Metaplasie in H
. pylori
infiziertem Patienten im Vergleich zu VacAs1 +, CagA + doppelt positiven Varianten [23]. Epidemiologisch Triple-positive Stämme mit der höchsten Inzidenz von Geschwüren und Magenkrebs korreliert sind [24].
Sialinsäure-bindenden Adhäsin (SabA)
Unabhängig von der Einhaltung fukosylierte Blutgruppenantigene über BabA, H
. pylori
bindet an Sialinsäure-modifizierten Glycosphingolipide, insbesondere Sialyl-Lewis-x /a (SLE X und sLeX a), über die bakterielle Adhäsin SabA [25]. Interessanterweise ist sLeX X fehlt in der gesunden, nicht-entzündeten Magenschleimhaut und damit SabA vermittelte Adhäsion wird ein relevanter Faktor bei der bakteriellen Persistenz nach erfolgreicher Kolonisierung und Etablierung von Entzündungsprozessen im Magen [25]. Dementsprechend Marcos und Kollegen [26] konnten zeigen, dass H
zu zeigen. pylori
-induzierten Entzündung führt zu einer erhöhten Expression der Glycosyltransferase β3GnT5, die in der Biosynthese der sLe X-Antigen als ein wichtiger Faktor wirkt. Die Induktion von β3GnT5 war auf Tumor-Nekrose-Faktor-alpha (TNF-α) abhängig, aber nicht IL-8, und Zellen ektopische β3GnT5 gab exprimieren höhere Haftung Raten für SabA-positive H
. pylori Stämme
[26]. Wie die Situation mit OIPA und BabA Expression von SabA unterliegt Schwankungen und Genkonversion in Phase mit seiner paralog SABB [27]. Zusätzlich Säure-responsive Signalisierung in H
. pylori
Grenzen SabA Transkription, die die H
anzeigt. pylori
Haftung ist ein dynamisches und regulierter Prozess [28, 29].
Adherence-assoziierten Lipoprotein A und B (AlpA /B)
Der OMP AlpA und AlPb wurden zunächst als Proteine beschrieben, die von H-Bindung erleichtern
. pylori
nach Kato-3-Zellen und der apikalen Oberfläche von Magengewebeschnitten [30, 31]. AlpA und AlPb teilen einen hohen Grad an Homologie und sind aus dem gleichen Operon co-transkribiert. Darüber hinaus sind beide Proteine für H
notwendig. pylori
vermittelter Adhäsion an Magenbiopsien [31]. Im Gegensatz zu anderen Adhäsine sind AlpA und AlPb nicht Variation der Phase unterworfen und praktisch alle klinischen Isolaten exprimieren sowohl Alp Proteine [32, 33]. Wichtig ist, dass fehlende Deletionsmutanten AlpA /B zeigten schwere Kolonisation Defekte in Maus und Meerschweinchen Tiermodellen [33, 34]. In scharfem Gegensatz dazu eine aktuelle Studie in der mongolischen Wüstenrennmäusen lässt vermuten, dass AlpA /B-defizienten Stämme zu überschwänglich Magenentzündung führen, wie zum isogenen gerbil angepassten Wildstammes verglichen [35]. Der Grund für diese widersprüchliche Ergebnisse in verschiedenen experimentellen Einstellungen, bleibt unklar.
Interessanterweise Lu et al. beschrieben in der Aktivierung von signifikanten Unterschiede Wege (Mitogen-aktivierte Proteinkinasen [MAPKs], c-Fos und c-Jun-, cAMP response element-binding protein [CREB] Signalisierungs -, Aktivatorprotein-1 [AP-1] - und NF-kappaB-bezogene Signalisierung), induziert durch H
. pylori
AlpA /B Deletionsmutanten [33]. Diese Daten implizieren, dass AlpA /B-vermittelte Adhärenz erleichtert eine stärkere Aktivierung bestimmter Signaltransduktionswege. Jedoch Injektion und Phosphorylierung von CagA sowie IL-8-Induktion wurden nicht signifikant beeinflusst durch AlpA /B Deletion [36]. H
. pylori
wurde Komponenten der extrazellulären Matrix (ECM), insbesondere Kollagen IV und Laminin [37], zu binden, gezeigt, die als in Frage kommende Wirtsfaktoren vorgeschlagen wurden, als Rezeptoren wirken. In diesem Zusammenhang wurde AlpA /B in der Adhäsion beteiligt an Laminin [35]. Als eine der Hauptkomponenten des ECM bindet Laminin an Integrin; Daher wäre es interessant zu untersuchen, ob AlpA /B indirekt Integrin Signalgebung durch die Bindung an Laminin modulieren können.
Äußere Entzündungsprotein A (OIPA)
OIPA auch auf die OMP-Gruppe gehört, und wurde vorgeschlagen, IL zu verstärken -8 Sekretion über Interferon-stimulierte responsive element (ISRE) wirkende parallel zur cag
PAI-abhängige Mechanismen [38, 39]. Dies steht im Gegensatz zu anderen Wieder Komplementation Studien darauf hinweist, dass in erster Linie OIPA Funktionen in H
. pylori
Adhäsion an Wirtszellen, während die IL-8-Ebene unberührt bleibt [36, 40]. Der Grund für diese gegensätzlichen Beobachtungen ist nicht klar.
Yamaoka und Mitarbeiter haben berichtet, dass die Expression von funktionellen OIPA in H
. pylori
phasenveränderlich und kann auf "ein" oder "aus" durch einen Fehlpaarung Mechanismus gerutscht Strang während chromosomalen Replikation ist [39, 41, 42]. Der OIPA Expressionsstatus wird oft mit dem Vorhandensein von cag assoziiert
PAI, VacAs1 und VacAm1 Allelvarianten in westlichen Typs klinischen Isolaten [40, 43, 44]. Daher ist es schwierig, relevante Korrelationen zwischen OIPA Status und klinische Manifestation zu liefern, für die OIPA Status vollständig von anderen krankheitsrelevanten genetischen Faktoren des Bakteriums, unabhängig zu sein scheint nicht.
Jedoch wie andere Adhäsine, OIPA erscheint ein wichtiger Faktor in der mongolischen Wüstenrennmaus-Infektionsmodell sein, da OIPA-defizienten Stämme gegen eine Infektion zu etablieren und nicht eine chronische Entzündung und Magen-Metaplasie [45, 46] induzieren. Bisher wurden keine spezifischen Rezeptor oder Oberflächenmolekül für OIPA Bindung wurde beschrieben. Trotzdem
, bezogen auf Infektionen mit einem OIPA
Deletionsmutante wurde OIPA vorgeschlagen Phosphorylierung von focal adhesion kinase (FAK) zu induzieren, was zu nachgeschaltete Aktivierung der MAPK extrazellulären signalregulierten Kinasen 1 und 2 (ERK1 /2) und die Bildung von Aktin-Stressfasern [47]. Zusammengefasst zeigen diese Daten, einen Wirtszellrezeptor mit der Fähigkeit, Signaltransduktion in Reaktion auf OIPA übertragen; Daher wäre es interessant zu untersuchen, ob rekombinante OIPA an einen Wirtszellrezeptor binden kann und FAK Signalgebung induzieren. Wie angedeutet durch einen genomischen Knock-out-Mutante, könnte OIPA-vermittelte Aktivierung FAK eine Folge der veränderten epidermalen Wachstumsfaktorrezeptor (EGFR) Signalisierung [47, 48] sein. Jedoch Aktivierung von EGFR wurde eine funktionelle T4SS zeugend gezeigt, benötigen [49] und rekombinante CAGL allein in der Lage, EGFR aktiviert [50]. Darüber hinaus wird ein OIPA
-knock-out-Mutante von H
. pylori
war, die EGFR-Signalkaskade beteiligt Phosphatidylinositol 3-Kinase (PI3K) → phosphoinositide-dependent kinase-1 (PDK1) → Akt auslösen nicht in der Lage, die an der Regulation der FoxO forkhead Transkriptionsfaktor-Aktivität wurde vorgeschlagen, beitragen [ ,,,0],51] und schließlich zur Induktion von IL-8-Sekretion [48]. In einer aktuellen Studie wurde vorgeschlagen, dass EGFR /FAK /Akt führt zur Phosphorylierung des focal adhesion protein paxillin signalisiert, die dann Zytoskelett-Reorganisation führt und anschließend Zellverlängerung [52].
Zusammengefasst ist OIPA eine interessante H
. pylori
Haftung Faktor, da es möglicherweise direkt mit Signaltransduktionswege stört, die durch Faktoren T4SS /CagA überwiegend aktiviert werden. Dies könnte darauf hindeuten, dass OIPA zu T4SS abhängige zelluläre Reaktionen beiträgt, entweder durch direkte Aktivierung eines noch nicht identifizierten Rezeptor oder indirekt durch enge Adhäsion zwischen H
vermitteln. pylori
und die Wirtszelle, was zu einer stärkeren T4SS /CagA-vermittelten Signal. In diesem Zusammenhang wäre es interessant zu untersuchen, ob die verfügbaren OIPA
Mutanten exprimieren noch voll funktionsfähig T4SS Pili.
Andere mutmaßliche Adhäsine Neben der gut beschriebenen Gruppe von Adhäsionsmoleküle
haben mehrere andere Faktoren gewesen in H
verwickelt. pylori
Adhäsion an die Magenschleimhaut. Der phasenvariable Protein HopZ ist vorgeschlagen worden, eine Rolle in der bakteriellen Adhäsion [53] und neuere Studien spielen konnte eine Rolle in der frühen Phase der Kolonisation zu demonstrieren. Re-Isolate von einem gesunden Freiwilligen herausgefordert mit HopZ 'off' H
. pylori
zeigte eine starke in vivo
Auswahl für den HopZ 'auf' Status [54]. Ein weiterer Bericht von Snelling und Mitarbeitern vorgeschlagen, eine haftungsbezogene Funktion für Horb [55]. Als zusätzliches OMP könnte HopQ auch einen Einfluss auf die bakterielle Adhäsion aufweisen. In einer Untergruppe von getesteten H
. pylori
Stämme, hopQ
Löschung erhöht H
. pylori
Einhaltung AGS-Zellen und führten zu einem hyperadherent Phänotyp und anschließend zu einer erhöhten Phosphorylierung CagA, während IL-8-Induktion nicht beeinträchtigt wurde [56]. Dementsprechend verringerte HopQ deutlich CagA-Injektion in Koinfektion Experimente in Magenepithelzellen [57]. Die Frage, ob HopQ stört die Funktion anderer Adhäsine in bestimmten H
. pylori Stämme
noch beantwortet werden. jüngsten Ergebnisse zeigen daher, dass ein HopQ knock-out Mutanten in einem anderen H
. pylori
Isolat hatte keinen Einfluss auf die bakterielle Adhäsion sind nicht unbedingt widersprüchlich. Die Expression von HopQ beigetragen
PAI-abhängige Signal und CagA-Injektion zu CAG, da diese durch hopQ
Wieder Ausdruck wiederhergestellt werden konnte [58]. Diese Daten legen nahe, dass H
. pylori
Adhäsine könnte auf zwei Arten handeln, entweder in einem kooperierenden oder in einer Maskierungs Weise.
H
. pylori
-secreted Urease, VacA und HtrA-: Faktoren in der Pathogenese Priming
Abgesonderte Faktoren ein hohes Potential aufweisen, da sie ganz am Anfang von mikrobiellen Infektionen wirken kann direkten Kontakt oder Haftung an den Wirtszellen ohne zu erfordern. In Sekretom Analysen von H
. pylori
, eine breite Palette von sekretierten oder extrazelluläre Faktoren identifiziert worden [59-61]. Obwohl die meisten extrazelluläre Proteine, die aus H
. pylori
weitgehend uncharacterized bleiben, unser Wissen von γ-Glutamyl-Transpeptidase (GGT), H
. pylori
Neutrophilen-aktivierendes Protein (HP-NAP), Urease, vakuolisierendes Cytotoxin A (VacA) und Hochtemperaturanforderung A (HtrA) stetig zunimmt. Zum Beispiel hat GGT in der löslichen Fraktion von H
identifiziert. pylori
[59] und wurde Kolonisierung von Mäusen zu verbessern gezeigt [62]. Interessanterweise können rekombinante GGT Apoptose und Zellzyklusarrest in AGS-Zellen induzieren [63, 64], aber der molekulare Mechanismus ist noch nicht geklärt. HP-NAP ist ein chemotaktischer Faktor H
. pylori
, dass in erster Linie zieht und aktiviert Neutrophile [65]; jedoch ist es nicht eine herausragende Rolle bei der Interaktion mit Epithelzellen spielen. Darüber hinaus haben verschiedene direkte Wirkung von Urease, VacA und HtrA- auf Magenepithelzellen beschrieben, einschließlich Induktion von Apoptose und geschwächt Integrität der interzellulären Adhäsionen (Abbildung 2b).
Urease
Die Urease-Komplex oft beschrieben worden ist, wie eine oberflächen präsentiert Virulenzfaktor von H
. pylori
. Die primäre Funktion der Urease Maschinen wird Pufferung des sauren pH von Harnstoff zu CO Umwandlung 2 und Ammoniak, die zur Neutralisierung der Magensäure um die Bakterien erforderlich ist. Es wurde lange angenommen, dass Urease sezerniert wird oder oberflächen lokalisiert und trägt wesentlich zur H
. pylori
's
Fähigkeit, in den Magen zu besiedeln und anhalten, da es tatsächlich ein säureempfindliches Bakterium zu sein [66] gilt. Die Bedeutung von Urease für erfolgreiche Besiedelung wurde in mehreren Studien hervorgehoben worden [66-68]; jedoch ein einzelner Bericht zeigt, dass Urease-negativen H
. pylori
Stämme sind noch in der Lage Mongolische Rennmäuse zu kolonisieren [69]., Die verschiedenen sequenzierten Genome von H
. pylori
ein Urease-Gencluster enthalten, die aus sieben konservierte Gene (UreA-B und E-I) besteht. Harnstoff- und UreB stellen die strukturelle Untereinheiten eines Ni 2 + -abhängigen hexameren Enzymkomplex. Urée UREF UREG und Ureh sind Hilfsproteine in Nickel Einbau beteiligt und Montage Enzym. Zusammen mit arginase ist UreI verantwortlich für eine nachhaltige Versorgung von Harnstoff unter sauren Umgebungsbedingungen [70]. Im Gegensatz zu der Hypothese von oberflächen lokalisierten Urease nimmt eine andere aktuelle Modell, dass die Haupt Ureaseaktivität in dem bakteriellen Zytoplasma befindet [71]. Neben seiner Rolle bei der erfolgreichen Kolonisierung H
. pylori
könnte Urease auch stören indirekt mit Wirtszellfunktionen. Urease-abhängige trägt Ammoniakproduktion um den Verlust von tight junction Integrität im Epithel, wie durch eine verminderte trans-epitheliale elektrische Widerstand (TEER) und verbesserte Occludin Verarbeitung und Internalisierung in in vitro Kulturen
gezeigt [72]. Offenbar Unterbrechung der tight junction Integrität war unabhängig von VacA und CagA in diesen Studien, die zu früheren Berichten in scharfem Kontrast ist [73, 74]. Die Wirkung von Urease auf Tight Junctions von einem anderen Bericht bestätigt, dass ureB
Löschung zeigt abrogiert H
. pylori
's
Fähigkeit tight junctions als CagA- oder VacA unabhängigen Prozess zu stören. Durch Regulieren der Myosin regulatorische leichte Kette-Kinase (MLCK) und Rho-Kinase scheint UreB Ausdruck für die Phosphorylierung von MLC [75] erforderlich werden. Auch wenn der genaue Mechanismus, durch die H
. pylori
Urease aktiviert dieser Signalweg bleibt unklar, können diese Daten erklären, wie Urease zu den Entzündungsreaktionen beiträgt, die die Störung der epithelialen Barriere begleiten.
vacuolating cytotoxin A (VacA)
Erste Anzeichen für ein sezerniertes Vakuole -induzierende Toxin wurde in Experimenten unter Verwendung von filtrierten H
gefunden. pylori
1988 Bouillonkultur [76]. Dieses Toxin wurde später als VacA identifiziert [77, 78]. Die zellulären Antworten auf VacA reichen von Vakuolisierung und Apoptose auf die Hemmung der T-Zell-Funktionen [79, 80]. Aufgrund dieser unterschiedlichen zellulären Antworten wird VacA als ein multifunktionales Toxin sein. Doch in den letzten Jahren ist es immer deutlicher, dass die meisten Effekte zurückzuführen sind auf die Anionen-Kanal in Abhängigkeit von VacA in mehreren Zellkompartimenten und verschiedenen Zelltypen. Innerhalb der Gensequenz, die Vielfalt der Signalsequenz (Allel-Typen s1 oder s2), Zwischenbereich (Allel-Typen i1 oder i2) und Mittelbereich (Allel-Typen m1 oder m2) beobachtet worden [81, 82]. Als Folge seiner mosaic Genstruktur ist das VacA-Protein sehr heterogen und existiert in verschiedenen Varianten mit Aktivitäten unterscheiden.
VacA als 140 kDa Protoxin mit einem N-terminalen Signalbereich ausgedrückt wird, einem zentralen Toxin-bildenden Region 88 kDa (p88) und eine C-terminale Autotransporterdomäne, die für die Sekretion des Toxins erforderlich ist [83]. Bei der Sekretion wird VacA weiter in zwei Untereinheiten verarbeitet werden, genannt VacA p33 und VacA p55 entsprechend ihrer jeweiligen Molekulargewicht, die Membran-Spanning Hexamere bilden [84, 85]. Es wurde vorgeschlagen, dass die VacA p55 Domäne für Zielzell-Bindungs [86] in erster Linie verantwortlich ist, während Vakuolisierung eine minimale Sequenz des gesamten VacA p33 und die ersten ~ 100 Aminosäuren von VacA besteht erfordert p55 [87, 88].
der genaue Mechanismus der VacA Eintritt in die Zielzellen noch trennend ist, durch die Tatsache, dass mehrere mutmaßliche Rezeptoren beschrieben worden. Dargestellt auf Epithelzellen, EGFR könnte als potentieller Kandidat dienen VacA zu binden, vor seiner Verinnerlichung [89, 90]. Ferner Rezeptor Protein-Tyrosin-Phosphatasen RPTPα [91] und RPTPβ [92] wurden als VacA-Rezeptoren beschrieben, die VacA-abhängige Vakuolisation fördern. Vaca Sphingomyelin in Lipid Rafts Bindung ist auch ein wichtiges Ereignis sein in VacA-vermittelte Vakuolisation [93] gezeigt worden. Im Gegensatz zu der Induktion von großen Vakuolen, fördert VacA ebenfalls die Bildung von Autophagosomen in Magenepithelzellen, die ihren Kanal-bildenden Aktivität erfordert [94]. Die Low-Density-Lipoprotein-Rezeptor-verwandtes Protein-1 (LRP1) als einen Rezeptor zu fungieren vorgeschlagen, die mit VacA interagiert autophagy und Apoptose [95] zu fördern. Weitere mutmaßliche Wirtszellrezeptoren für H
. pylori
VacA vorgeschlagen worden; Es bleibt jedoch unklar, ob sie als echte Rezeptoren funktionieren. Da es nicht klar ist, ob identifiziert VacA Rezeptoren unabhängig voneinander funktionieren, impliziert die Identifizierung eines solchen Vielfalt an Rezeptoren ein komplexes Netzwerk von Interaktionen und konnte die pleiotropen Funktionen H
zugewiesen erklären. pylori
VacA. pylori
Infektionen. Alle Autoren gelesen und genehmigt haben das endgültige Manuskript.
 Das Darmmikrobiom ist auch im fetalen Leben Realität
Das Darmmikrobiom ist auch im fetalen Leben Realität
 Darm- und orale Mikrobiome sagen den Schweregrad von COVID-19 voraus
Darm- und orale Mikrobiome sagen den Schweregrad von COVID-19 voraus
 Darmbakterien im Zusammenhang mit stärkeren Muskeln bei älteren Menschen
Darmbakterien im Zusammenhang mit stärkeren Muskeln bei älteren Menschen
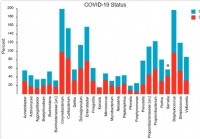 Zusammensetzung und Struktur des nasopharyngealen Mikrobioms beziehen sich auf die Schwere der COVID-19-Krankheit
Zusammensetzung und Struktur des nasopharyngealen Mikrobioms beziehen sich auf die Schwere der COVID-19-Krankheit
 Studie deutet auf Zusammenhang zwischen probiotischem Konsum und „Gehirnnebel“ hin
Studie deutet auf Zusammenhang zwischen probiotischem Konsum und „Gehirnnebel“ hin
 Das Lungenmikrobiom sagt den Schweregrad der COVID-19-Krankheit voraus
Das Lungenmikrobiom sagt den Schweregrad der COVID-19-Krankheit voraus
 Milchsäurebakterien und Darmbakterien tragen zu den gesundheitlichen Vorteilen von Roggen bei,
Studie zeigt Der Verzehr von Roggen hat eine Vielzahl von gesundheitlichen Vorteilen. Eine neue Studie der University of Eastern Finland zeigt nun, dass sowohl Milchsäurebakterien als auch Darmbakteri
Milchsäurebakterien und Darmbakterien tragen zu den gesundheitlichen Vorteilen von Roggen bei,
Studie zeigt Der Verzehr von Roggen hat eine Vielzahl von gesundheitlichen Vorteilen. Eine neue Studie der University of Eastern Finland zeigt nun, dass sowohl Milchsäurebakterien als auch Darmbakteri
 Darmbakterien im Zusammenhang mit Stoffwechselveränderungen und Autismus in neuer Studie
Forscher des California Institute of Technology haben eine wichtige Entdeckung gemacht, die erklären könnte, wie Darmbakterien zu autismusähnlichem Verhalten beitragen können. Das Team fand heraus,
Darmbakterien im Zusammenhang mit Stoffwechselveränderungen und Autismus in neuer Studie
Forscher des California Institute of Technology haben eine wichtige Entdeckung gemacht, die erklären könnte, wie Darmbakterien zu autismusähnlichem Verhalten beitragen können. Das Team fand heraus,
 Neugeborenes Mausmodell liefert Hinweise auf die Ursache einer verheerenden Darmerkrankung bei anämischen Frühchen
Ärzte haben seit langem den Verdacht, dass durch Transfusionen von roten Blutkörperchen bei Frühgeborenen mit Anämie die Gefahr der Entwicklung einer nekrotisierenden Enterokolitis besteht. oder NEC,
Neugeborenes Mausmodell liefert Hinweise auf die Ursache einer verheerenden Darmerkrankung bei anämischen Frühchen
Ärzte haben seit langem den Verdacht, dass durch Transfusionen von roten Blutkörperchen bei Frühgeborenen mit Anämie die Gefahr der Entwicklung einer nekrotisierenden Enterokolitis besteht. oder NEC,