Identifiering av DNA-metylering förändringar i samband med human magcancer Bild Sammanfattning
Bakgrund
Epigenetisk förändring av genuttrycket är en vanlig händelse i human cancer. DNA-metylering är en välkänd epigenetiska process, men kontrollera den exakta innebörden av epigenetiska förändringar i samband med cancer är fortfarande svår.
Metoder
Vi profilerade den methylome av human magcancer vävnad vid 50 bp upplösning med hjälp av en metylerad DNA anrikning teknik (metylerad CpG-ö återhämtning analys) i kombination med en genom analysator och en ny normalisering algoritm.
Resultat
Vi kunde få en heltäckande bild av promotorer med olika CpG densiteter, inklusive CpG-öar (CGI), avskrift organ och olika upprepnings klasser. Vi fann att magsäckscancer var associerad med hypermetylering av 5 'CGI och 5'-änden av kodande exoner samt hypometylering upprepade element, såsom korta inblandade kärnelement och kompositelementet SVA. Hypermetylering av 5 'CGI var signifikant korrelerad med nedreglering av associerade gener, såsom de i HOX Köpa och histon genfamiljer. Vi upptäckte också långväga epigenetiska tyst (LRES) regioner i gastric cancervävnad och identifierat flera hypermethylated gener (MDM2
, DYRK2
och LYZ
) inom dessa områden. Metylering status CGI och gen anteckning element i metastaserande lymfkörtlar var ett mellanting mellan normal och cancervävnad, vilket tyder på att metylering av specifika gener ökas gradvis i cancervävnad.
Slutsatser
Våra resultat kommer att ge värdefulla data för framtida analys CpG metylering mönster, användbara markörer för diagnos av magcancer, samt en ny analysmetod för kliniska Epigenomics undersökningar.
bakgrund
Gastric cancer är den näst vanligaste orsaken till cancer dödsfall i världen efter lungcancer, vilket resulterar i mer än 800.000 dödsfall i världen varje år [1]. Den nuvarande 5-års överlevnad på individer med diagnosen magcancer är endast 20-30%, med detta låg kan tillskrivas det faktum att de flesta fall redan i ett framskridet stadium när diagnosen. Som i alla cancerformer förblir tidig upptäckt den mest lovande strategi för att förbättra överlevnaden. Därför att förstå orsaken till tumörbildning i human gastrisk vävnad är viktigt.
Infektion med H. pylori
är en väletablerad och vanligaste orsaken till magcancer. Men förändringar i olika genetiska faktorer är också viktiga för att öka gastrisk cancerrisken. Det är väl känt att kromosomal instabilitet härrör från genetiska faktorer, såsom mikrosatellitinstabilitet samt KRAS
och p53
mutationer resulterar i utvecklingen av tumörer. Flera genomiska studier har identifierat nedärvda mutationer i specifika gener [2-4] och sjukdomar mottagliga loci [5, 6] för magcancer. Nyligen genomförda studier som jämför magcancer och normal vävnad har identifierat ett antal genetiska markörer, inklusive diagnostiska markörer [NF2
[7], INHBA
[8], SFRP4
[9]], prognostiska markörer [CD9
[10], CDH17
[11], PDCD6
[12]], och magcancer associerade gener [MUC13
[9], CLDN1
[13], Ki67
och CD34
[14]]. Dessutom har epigenetiska mekanismer såsom DNA-metylering och histon ändringar visat sig vara viktiga för att reglera uttrycket av gener som är involverade i biologi och sjukdomar i mag-tarmkanalen [15].
DNA-metylering spelar en viktig roll i eukaryoter och förknippas med ett antal nyckel mekanismer, inklusive genetisk prägling, X-kromosom inaktive, åldrande och cancer. Förändring av DNA-metylering i genomet finns i nästan alla typer av cancer och kan leda till förändringar i genuttryck, till exempel överexpression av onkogener och tystande av tumörsuppressorgener under cancerutveckling [16]. Flera studier har visat att ackumulering av genetiska och epigenetiska förändringar i gastriska precancerous lesions kan påverka ett stort antal mål, såsom DNA-reparationssystemkomponenter, tumörsuppressorer, onkogener, cellcykelregulatorer, tillväxtfaktorer och adhesionsmolekyler [17-20] . Emellertid har dessa studier varit främst inriktad på ett fåtal gener eller täckas endast en del av hela genomet. Således, tillgång till ett globalt perspektiv på de epigenetiska förändringar i samband med cancerutveckling har varit svårt. I synnerhet förståelse DNA metylering förändringar i intragenic regioner, CpG-öar, intergena regioner och upprepa sekvenser fortfarande begränsad. Följaktligen finns det ett stort intresse i genomet hela analys av avvikande DNA-metylering i dessa regioner. Idéer för omfattande genomet skala profilering av DNA-metylering i embryogenes och carcinogenes, högupplösta hela genomet sekvenseringsmetoder såsom BS-artiklar [21 -24], har MeDIP-artiklar [25, 26], och MethylCap-artiklar [27-29] utvecklats. Trots den snabba utvecklingen av sekvenseavbildningsteknik, finns det fortfarande en brist på jämförande forskning, vilket är avgörande för kliniska Epigenomics studier, inklusive sådana som är inriktade på cancer. Till skillnad från microarray-baserade metoder är sekvenseringsdata fram i ett format som inte är mottaglig för differentialanalys och analys arbetsflöde har inte standardiserats. Därför är beräknings billiga normaliseringsmetoder som behövs för att hantera beräkningsbördan att behandla stor storlek, hög upplösning sekvenseringsdata.
Här införde vi en normalisering algoritm, som tar hänsyn till provspecifika totala läsning densitet, den rumsliga fördelning av CpG loci, och bakgrundssekvense partiskhet. Vi skapade sedan en omfattande hel-genomet methylome av normal gastrisk vävnad, magsäckscancer vävnad, och metastaserande lymfkörtlar med hjälp av MethylCap-punkter metod och fick detaljerad information om sin störning under karcinogenes och metastas. Detta är lätt tillämpas på en jämförande analys av methylomes och andra typer av epigenomiska uppgifter, och det har särskilda konsekvenser för kliniska epigenomik.
Metoder
Gastric vävnadsprover
vi fått tre gastriska tumörer snabbfrystes och matchade normala gastric vävnad från Seoul National University College of Medicine för methylome studie. Dessutom var tjugoåtta matchade par av normala och tumör magen vävnader som erhållits för ytterligare bekräftelse. Alla prover erhölls genom endoskopisk resektion vid undersökning av de patienter som gav informerat samtycke
metylerat DNA återhämtning analys (MIRA) Review Genomisk DNA från 25 mg av gastric vävnad renades med DNeasy Blood &. Vävnads Kit (Qiagen, Valencia, CA). Genomiska DNA-prover från 3 individer poolades vid samma koncentration. MIRA utfördes såsom tidigare beskrivits [30-32]. Kortfattat, GST-märkt MBD2b och His-märkt MBD3L1 proteiner framställdes såsom beskrivits. 15 ug av genomiskt DNA fragmenterades till 100 ~ 500 bp genom sonikering och inkuberades med 28 ug av renat GST-MBD2b protein, 28 | ig His-MBD3L1 protein och 7 ug JM110 Bakteriell DNA under 6 timmar. 30 ul av MagneGST pärlor (Promega, Madison, WI) förblockerades med 7 | ig av JM110 bakteriellt RNA tillsattes och inkuberades vid 4 ° C med roterande under 45 min i slutlig 600 ul av MIRA-bindningsreaktionsblandningen. Kulorna tvättades tre gånger med 1 ml tvättbuffert, och metylerade-fragment eluerades genom inkubation vid RT under 5 minuter och därefter 56 ° C under 30 minuter med 30 ul av TE innehållande RNas A (100 | ig, Qiagen) och proteinas K ( 15 ug, Qiagen). Eluerade DNA-fragmenten renades ytterligare genom att använda Qiaquick PCR-reningssatser (Qiagen).
Illumina Genome Analyzer sekvense Review Vi använde 10 ng av eluerat DNA för Illumina Genome Analyzer sekvensering. Efter ligering av ett par av Solexa adaptrar, ligeringsprodukter med den maximala insertstorlek på 200 bp gelrenades på 2% agaros och utsattes för PCR-amplifiering. Kluster produktions- och 36 cykler av sekvensering utfördes genom att följa tillverkarens instruktioner. Vi sekvens 120 ul av adapter-ligeras, storleksfraktion DNA (2 ~ 04:00) på Illumina Genome Analyzer. Sekvens taggar kartlades till det mänskliga genomet (UCSC hg18 databas baserad på NCBI Build 36,1 montering) med Solexa Analys Pipeline (version 0.3.0). Sekvenserades läser av 34 bp (exklusive den första och sista nukleotid) som passerade kvalitetskontroll filter användes. Bearbetning
Data och MES beräkning
Vi förlängde 3 'änden av 34-bp läser av 200 bp för att täcka DNA fragment bundna av MBD proteiner. Avläsningen omvandlades till webbläsaren töjbara data (BED) filer för visualisering i UCSC genomet webbläsare http: //. Genomet ucsc edu /.. Vi räknade överlappande sekvenstaggar vid 50 upplösning bp. Att hitta anrikade genomregioner, räknades antalet mappade läser in ett glidande fönster av en kb jämfört med det totala antalet läser eller bakgrunden antalet läser i genomet. Som sådan var MES beräknas på två sätt; en är som log2 av (mål läs count /målstorlek) /(total read count /genomstorlek) och täckt med noll, den andra är som log2 av (mål läs count /målstorlek) /(bakgrund läs count /bakgrund storlek) och täckt med noll. Att justera för bakgrundssekvense partiskhet, var MESbg beräknas på samma sätt för inmatning sekvense utan affinitetsrening och subtraheras från MES.
Genomic ståndpunkter CGI, promotorer, avskrift kroppar, CDS, och repetitiva element | Allt iska ståndpunkter CGI, avskrifter och upprepa element hämtat från UCSC genomet webbläsare. Totalt 27.639 CGI (utom slumpvis belägna CGI) förutsågs av följande kriterier: GC-halt av 50% eller mer, längd större än 200 bp, och förhållandet är större än 0,6 av observerade antalet CpG-dinukleotider till det förväntade antalet [33] . NCBI mRNA referenssekvenserna samling (RefSeq från versionen 46, 11 mars 2011) användes för att identifiera transkriptionsenheter med den definierade transkriptionsstart, end platser och CDS start, slut platser. För promotorer använde vi regionen 500 bp uppströms ~ 500 bp nedströms om transkriptionsstartstället. Vi fick ~ 5 miljoner återkommande platser som hade fastställts av den RepeatMasker program baserat på RepBase bibliotek upprepningar.
Metylering nivå iska element
metylering nivån en CGI, promotor, gen-organ, och upprepa elementet uppskattades med hjälp av MES som överlappar varje element. MES = 0 användes för att definiera ometylerade element. För att mäta hypermethylation eller hypometylering i cancer, vi beräknade differential röra som (Cancer MES - Normal MES). Differential MES > 1,0 användes som en tröskel. För att förstå de funktioner utvalda gener, använde vi ontologi klassificering av gener genom DAVID Funktionell Notering Clustering verktyg http:.... //David ABCC ncifcrf gov /News genexpressionsanalys
microarray produkt som används i denna studie var Codelink Human Whole Genome 55 K chip (GE Healthcare, USA). Alla experimentella procedurer inklusive förberedelse cRNA mål, hybridisering, var efter hybridisering färgämne koppling utförs med hjälp av leverantörs rekommenderade protokoll. De resultatfiler importerades till GeneSpring GX 7,3 (Agilent Technologies, USA) för filtrering och grundläggande statistisk analys. Bland 55 K gener på microarray, bara gener med nuvarande flaggor i åtminstone 50% av proverna ut för efterföljande analys. Microarray data avsattes på GEO http:.. //Www NCBI NLM NIH gov /geo /(åtkomstnummer GSE33651) Review MIRA och realtid qPCR
MIRA... utfördes på ytterligare fyra individuella prover. DNA renades från supernatanten och övervakas av realtid qPCR hjälp av Roche 480 maskin. Sekvenserna för begagnade primers presenteras i kompletterande fil 1:. Tabell S1
bisulfit behandling, metylering-specifik PCR och pyrosekvensering
Vi isolerade det genomiska DNA från individ prov genom användning av en Qiagen DNeasy Tissue Kit (Qiagen). Bisulfit behandling genomfördes med hjälp av EZ DNA-metylering guld kit (Zymo forskning) enligt tillverkarens anvisningar. Bisulfit-behandlad DNA vid -80 ° C tills vidare användning. De primers som användes för MSP utformades med hjälp av Methprimer [34], och visas i kompletterande fil 1: Tabell S1. PCR utfördes med HotStarTaq Polymerase (Qiagen) och inkluderade en initial inkubering vid 95 ° C under 15 min, följt av 40 cykler av 95 ° C under 1 min, 59 ° C under 1 min och 72 ° C under 40 sekunder, följt av en cykel av 72 ° C under 10 minuter. MSP produkter separerades på 2% agarosgeler och visualiserades genom EtBr färgning. Pyrosequencing reaktionerna utföras automatiskt med ett PSQ 96 System (Pyrosequencing AB) enligt tillverkarens anvisningar. I korthet var den biotinylerade PCR-produkten (50 ul) renades genom användning av streptavidin-sepharose pärlor (Amersham Biosciences). Den renade produkten laddas in i reagenspatronen med enzymet, substratet och dNTP inkluderas i PSQ96 SNP Reagent Kit (Pyrosequencing AB). Sekvense primers för Pyrosequencing visas i kompletterande fil 1. Tabell S1
Resultat
Bearbetning av MIRA-punkter methylome uppgifter
Vi renade metylerat DNA berikas genom MIRA (metylerad CpG-ö återhämtning analys) och sekvenserat DNA med nästa generations sekvensering. DNA-metylering nivåer bestämdes med användning av sekvense läsa räkningar av motsvarande regioner, på 50 bp intervaller, såsom beskrivits under metoder. Vi skapade DNA-metylering kartor för både normala och cancer mag vävnader. För varje prov fick vi cirka 10 miljoner sekvens läser (Ytterligare fil 1: Tabell S2). Varje methylome innehöll ~ 140 miljoner CpG läser, som täcker ~ 48% av alla genomiska CpG platser exklusive centromerer (Ytterligare fil 1: Tabell S3). Den genomsnittliga täckningen av CpG läser i varje methylome var 4.5X. Till stöd för den höga känsligheten hos MIRA, genomsegment som endast innehåller en CpG hade högre läsa räkningar än de utan CpG (p
värde = 0), vilket tyder på att enstaka CpG förändringar skulle kunna lösas med hjälp av MIRA. Den genomsnittliga sekvens läser ökat i proportion till antalet CpG inom en 50-bp intervall, och i själva verket MIRA täckning var inte låg, även för regioner med låg CpG densitet (Ytterligare fil 2: Figur S1). Sammantaget visar dessa resultat att MIRA var framgångsrik i att återvinna en tillräcklig fraktion av metylerade regioner. Som för riktigheten i MIRA, ~ 99% av MIRA-tagna fragment hade åtminstone en CpG plats inom sin sekvens, vilket tyder på en låg falsklarm ränta.
Att mäta anrikning av lokala metylering signaler, vi beräknade metylering anriknings poäng (MESs ) genom att erhålla en läsa räkning i en viss region och sedan utföra normalisering för att styra det totala läsa räkningen (MEST) i provet (global normalisering) eller den lokala läs räkningen (MESl) i en användardefinierad omgivande regionen (lokal normalisering) (se Metoder). Detta möjliggör en direkt jämförelse av oberoende prover med olika läs densitet. Vi genomförde därefter en logaritmisk omvandling av det erhållna poäng. Tillsammans med att ha andra matematiska meriter, ger detta fördelen variansstabilisering, särskilt för hög lästa räknas, som ofta är kopplade till höga tekniska variationer som kan införa betydande partiskhet i data.
Vi bedömde den statistiska signifikansen av MES i två sätt. Randomiserad MESs genererades numeriskt genom att permutera de iska positioner av vår sekvens läser. Bakgrunds MES (MESbg) experimentellt erhållits genom sekvensering av normala genomet utan affinitetsrening. Som väntat, de verkliga data gav betydligt högre anriknings poäng (Ytterligare fil 2: Figur S2). Noterbart MESbg var högre än MES från randomiserade genom, en indikation på att bakgrundssekvenser ensam kan skapa anrikning, förmodligen på grund av kromatin tillgänglighet och förstärkning partiskhet. I överensstämmelse med de senaste rapporterna [35], visar detta att det behövs en korrekt kalibrering för inneboende sekvense partiskhet. Därför normalise vi våra MES med MESbg.
Att hitta den optimala villkor för normalisering, vi jämförde statistiska lämplighet olika normaliseringsmetoder. Tag fördelning längs genomet kan modelleras av Poisson fördelning [36, 37]. Godhet passar testades med användning av Kolmogorov-Smirnov test. I detta test indikerar en låg D statistik en bra passform. Medan Poisson modellen bättre än Gauss övergripande, MES visade en bättre passform än råa läs räknas (Ytterligare fil 2: Figur S3), som visar de sällsynta fall naturen av logg skalas läsa räkna åtgärd. De normaliserade MESl kalibrerade av styrsekvensering (MESbg) gav ännu bättre resultat än normaliserad MEST kalibreras av styrsekvensering (MESbg) Global. Mössor och kromosomala utsikt över DNA-metylering
efter bekräftar den bästa metoden för poängsättning genomomfattande metylering nivåer på 50-bp intervaller undersökte vi de kromosomala metyleringsmönster av normala prover. Den genomsnittliga MES beräknas för varje kromosom föreslog att CpG-rika och gen-rika kromosomer tenderar att vara mycket metyleras (Figur 1A). Metylering nivåer av kromosomer med stora mängder lång insprängda kärnelement (linjer) var relativt låg (t ex kromosom 4). Interestingly, mängden korta inblandade kärnelement (SINES) var proportionell mot kromosom metylering mönstret. Detta är sannolikt orsakas av det faktum att sinus typiskt grupperade i gen-rika regioner. Könskromosomer var globalt hypomethylated med lägre CpG densitet och högre upprepa innehåll än autosomer. Eftersom vi använde vävnader som tas från en hane i detta experiment, är den globala hypometylering av X-kromosomen observerade inte förknippas med X inaktivering. utsikt kromosomomfattande sammanfattat hög CpG densitet och hög metylering runt gen-rika (se svarta fält längst ner) och CGI-rika regioner (se blå staplar längst upp) (Figur 1B). Däremot var låg CpG densitet och låg metylering observeras runt gen fattiga regioner som var rik på långdistans upprepningar (> 1 kb) (se röda staplar längst upp). De genomsnittliga MES tyder på att metylering nivån av CGI är avsevärt högre än den för gena regioner eller upprepningar (Figur 1B). Figur 1 metyleringsmönster av normal gastrisk vävnad. (A) Kromosomomfattande genomsnittliga MES visas som en funktion av den genomsnittliga CpG densitet, gen densitet (antalet gener per Mb), LINE kvantitet (längden på LINE per Mb), och SINE kvantitet (längden på SINE per mb) för varje kromosom. (B) För kromosom 22, var den genomsnittliga CpG densitet (skuggade) och MES (svart kurva) som erhållits i en-Mb skjutfönster. Positionerna för transkriberade gener (svarta staplar längst ned), CG öar (blå staplar längst upp), och långa upprepningar (> 1 kb, röda staplar på toppen) jämförs mot bakgrund av DNA-metylering och CpG densitet ( vänster). Den genomsnittliga MES för CGI, gen organ, och upprepar (höger). (C) Fördelningen av genen organ och CGI MES (vänster). Den genomsnittliga MES för promotor-associerade och promotor oberoende CGI visas till höger. (D) Den genomsnittliga MES för promotorundergrupper, baserat på förekomsten av CGI (till vänster). (E) Grundläggande information om intergena, exonic och intronregioner, enligt längd, CpG-nummer, och mappas läser (vänster). Fördelningen av intergena, exonic och intron MESs visas till höger. (F) Grundläggande information om uppströms en-kb region 5 'UTR exoner kodande exoner, 3' UTR exoner och nedströms en-kb region enligt längd, CpG nummer och mappade läser (vänster). Fördelningen av MES för varje element visas till höger.
Allmänhet CGI tenderar att förbli metylering gratis i normal vävnad. Att analysera hög metylering mönster av CGI, vi kontrollerat den genomsnittliga MES distribution och fann en något bimodal mönster (Figur 1C). Cirka 66% (11.376 /17.284) av CGI i den vänstra toppen överlappade med en promotor (1 kb från vår definition). Däremot 13% (1386 /10.357) av CGI i rätt toppen överlappade med en promotor, vilket tyder på att de flesta promotor associerade CGI är ometylerade. I motsats till promotor relaterade CGI, promotor oberoende CGI var kraftigt metylerat (Figur 1C). Även om de flesta CGI-positiva promotorer inte denaturerad, CGI-negativa promotorer visade relativt höga metylering nivåer (figur 1D). Vi kontrollerade också metylering nivån promotorer av CpG densitet enligt tidigare definition [38] (Ytterligare fil 3: Tabell S4). Metylering mönster av promotorer var omvänt relaterad till CpG densitet (Ytterligare fil 2: Figur S4). Å andra sidan, CGI-innehållande gen kroppar hade högre metylering nivåer än de utan CGI (figur 1D).
Därefter analyserade vi metylering anrikningsmönster vid olika kommenterade genom element för att utforska områden som företrädesvis metylerad. Gena regioner upptar cirka 40% av människans arvsmassa, men ungefär 53% av den totala läser är inom detta område, med de flesta läser är belägen i intronregionen (tabell 1). Även om en betydande del av metylerade fragment faller inom intronregioner, förhållandet mellan mappade läser till längden på exon är betydligt högre än för introner, vilket tyder på att exoner är högre metylerade än introner (Figur 1E). Inom gen-relaterade områden, är anrikningen av kodande exoner och med högre än i andra regioner som tidigare rapporterats (Figur 1F och tabell 2) [39]. Detta tyder starkt på att metylering spelar en roll i exon regulation.Table en mänskliga genomet och normal prov information om framkallande och intergenregion
Human Genome Information
Normal prov information
Relativ Anrikning Ratio
Funktionell Kategori
Längd (bp)
Ratio
# av CpG
förhållande
Läser
förhållande
vs. längd
vs. CpG Count
Genic
1,184,139,094
39.46
13,262,253
47.09
20,854,434
53.25
1.35
1.13
Exon
68,035,894
2.27
1,808,089
6.42
4,350,405
11.11
4.90
1.73
Intron
1,122,817,725
37.41
11,613,113
41.23
17,358,273
44.32
1.18
1.07
Intergenic
1,816,976,186
60.54
14,901,610
52.91
18,310,273
46.75
0.77
0.88
Human Genome
3001115280
100
28.163.863
100
39.164.707
100
1,00
1,00
Tabell 2 mänskliga genomet och normal prov information om gen kommenterade regioner
Human Genome Information
Normal prov information
Relativ Anrikning Ratio
Funktionella kategori
Längd (bp)
Ratio
# av CpG
Ratio
Läser
Ratio
vs. längd
vs. CpG Count
Upstream 1 kb
24.468.069
0,82
937.748
3,33
535.593
1,37
1,68
0,41
5'UTR Exoner
8436529
0,28
411563
1,46
292654
0,75
2,66
0,51
kodande exoner
33384619
1,11
1077913
3,83
3448755
8,81
7,92
2,30
3'UTR Exoner
28387978
0,95
378012
1,34
806832
2,06
2,18
1,53
Nedströms en kb
23.136.263
0,77
340.866
1,21
551.071
1,41
1,83
1,16
Human Genome
3001115280
100
28163863
100
39164707
100
1,00
1,00
Förändringar i DNA-metyleringsmönster associerade med magcancer
När genomsnittliga kromosom MES av cancer methylome jämfördes med den för kontrollvävnad, fann vi att alla kromosomer i cancervävnaden tenderade att hypomethylated (Ytterligare fil 2: Figur S5). Med utsikt kromosomomfattande var CGI-rika regioner visade sig vara specifikt hypermethylated, medan upprepade rika regioner allmänt hypomethylated (Figur 2A, ytterligare fil 2: Figur S6). För att analysera metylering förändringar i iska element, i linje vi varje element vid start- och slutplatser och sedan erhålls de genomsnittliga MES vid respektive position. Påfallande upptäckte vi hypermethylation i uppströmsregionen, i synnerhet från 500 bp uppströms om transkriptionsstartstället (figur 2B). Detta är i enlighet med den hypermetylering av promotorregioner ofta observerats i cancer. Figur 2 Jämförelse av metyleringsmönster i normal och cancerös vävnad. (A) Genomsnittlig MES kurvan för normal (svart) och cancer (röd) vävnad i kromosom 19 (till vänster). Den genomsnittliga MES för CGI, gen organ, och upprepningar (höger). (B) DNA-metylering av genen kommenterade element. Varje element (uppströms en kb, exon, intron, och nedströms en kb) fördelades i 20 fack och den genomsnittliga MES erhölls för varje fack av alla motsvarande element. (C) DNA-metylering vid transkript ändarna och den kodande regionen ändar. De genomsnittliga MES erhölls i ett glidande 50-bp fönstret enligt dess avstånd från transkriptet start (första) och slutet (andra) för CGI-positiva promotorer samt transkriptet start (tredje) och slutet (fjärde) för CGI- positiva promotorer. (D) DNA-metylering av totalt 5 'UTR exoner (vänster) och 5' UTR-kodande exoner (höger).
Region centrerad vid transkriptionsstartstället visade helt olika mönster beroende på närvaron av ett CGI, vilket avspeglar den låga metylering status CGI innehåller promotorer (figur 2C). Vi fann också att, i cancervävnad, sker anmärkningsvärd hypermetylering av CGI-innehållande promotorer och att densiteten av CpG är avgörande för ökningen av DNA-metylering (figur 2C). För att ytterligare analysera huruvida 5 'regioner gener hypermethylated likhet med genpromotorer, vi kontrollerat metylering mönstret av de första exonerna. Intressant nog fann vi att den första exonen var hypermethylated endast när det var 5'-änden av en kodande exon, men inte när det var en 5 'UTR exon (figur 2D). Dessa regioner innehåller också hög CpG densitet. Därför CGI vid uppströms regionerna av gener, promotorn och kodnings start verkar vara de viktigaste målen för DNA hypermethylation i cancer.
Metylering mönster av CpG-öar
Att undersöka sambandet mellan placeringen av CGI och DNA-metylering, subgrouped vi CGI enligt deras position inom genomet. Specifikt var de kategoriseras som 5 '(belägen mellan en kb uppströms och den kodande startstället för en gen), intragenic (intragenic CGI utanför 5'-änden), och intergena (finns i icke-gena region) (Ytterligare fil 1: tabell S5). Även CpG densitet var liknande bland de tre grupperna, icke-5 "CGI (intragenic och intergena CGI) var betydligt mer metylerade än 5 'CGI (Ytterligare fil 2: Figur S7). Vi jämförde vidare de genomsnittliga MES av subgrouped CGI och fann metylering av alla CGI i allmänhet ökat. Men den relativa differentiella MES föreslog att förändringen i metylering i 5 'CGI var betydligt större än för andra CGI (figur 3A), vilket återspeglar de viktiga roller 5' CGI i cancer. Omfattningen av 5 'CGI hypermetylering korrelerade signifikant med överlappningen av transkriptionsstartstället (figur 3B). Figur 3 DNA-metylering av CpG-öar. (A) Relativ differential MES av subgrouped CGI. (B) Samband mellan differential CGI metylering och avståndet till transkriptionsstartplatsen. (C) Samband mellan genuttryck nivå och hypermetylering av CGI. (D) metylering specifika PCR av histon gener som visar de högsta differential MES värden. . M1 och U1 motsvarar HIST3H2A, medan M2 och U2 motsvarar HIST3H2B
att utforska funktionen av gener som genomgår differentiell metylering vid 5 'CGI, valde vi gener med högt differential CGI MESs (differential MES > 1). Vi har sedan utfört gen ontologi (GO) analys för att få insikt i de mekanismer som är ansvariga i cancer (tabell 3). När generna klustrade i olika GO kategorier, fann vi att HOX
genkluster och nukleosomen monteringsrelaterade genkluster var mål för hypermetylering, medan apoptosrelaterade genkluster var mål för hypometylering. Intressant nog är vår slutsats att HOX
genkluster var förmånliga mål för DNA-metylering i linje med en tidigare rapport [40]. Dessutom bekräftade gen tomter som hypermetylering var CGI-specifik cancer (Ytterligare fil 2: Figur S8). För att uppskatta förändringar i uttrycksmönster som orsakas av hypermetylering av 5 'CGI, utförde vi en funktionell analys av genuttryck data som erhållits från cDNA microarray experiment. Hypermetylering av 5 'CGI var signifikant korrelerad med nedreglering av gener (p
= 0,03) (Figur 3C, ytterligare fil 3: Tabell S6 och S7). Detta tyder på att tysta gener genom metylering kan påverkas direkt av graden av CpG densitet och 5 'CGI hypermethylation. Vi analyserade DNA-metylering status av gener med hypermethylated 5 'CGI och nedreglerade uttrycksmönster. Bland dessa var den gen som kodar histon H2B typ 3-B (HIST3H2BB
). Analys av HIST3H2BB
promotor metylering med användning av metylering-specifik PCR visade att de flesta cancerpatienter (10/08, 80%) uppvisade ökad metylering i promotorregionen (figur 3D) .table 3 Funktionell annotering klustring av gener med hypermethylated 5'CGIs
Notering Cluster en
Anrikning Poäng: 3,27
Count
P_Value
GOTERM_BP_FAT
nukleosomen montering
11
3.90E-04
GOTERM_BP_FAT
kromatin montering
11
5.20E-04
GOTERM_BP_FAT
protein DNA-komplex montering
11
7.40E-04
Notering Cluster 2 Review Anrikning Poäng: 2,92
Count
P_Value
Interpro
Histon kärna
8
6.80E-04
SP_PIR_KEYWORDS
nukleosomen kärna
8
8.60E-04
GOTERM_CC_FAT
nukleosomen
8
3.10E-03
Notering Cluster 3
Anrikning Poäng: site
17
5.10E-03
INTERPRO
Homeobox
17
5.70E-03
SP_PIR_KEYWORDS
Homeobox
17
5.80E-03
INTERPRO
Homeodomain-related
17
6.40E-03
SMART
HOX
17
1.40E-02
D4
3
9.10E-03
SP_PIR_KEYWORDS
embryo
3
3.30E-02
PIR_SUPERFAMILY
PIRSF002612:homeotic
 Bästa GI-läkare i USA/ Högst rankade gastroenterologer – Dr. Vikram Tarugu
Om du är redo att lära dig om de bästa Gatroenterologerna i USA som ger de mest effektiva behandlingarna så läser du rätt guide. Vi har sammanställt denna korta men exakta guide för att ge dig omfatta
Bästa GI-läkare i USA/ Högst rankade gastroenterologer – Dr. Vikram Tarugu
Om du är redo att lära dig om de bästa Gatroenterologerna i USA som ger de mest effektiva behandlingarna så läser du rätt guide. Vi har sammanställt denna korta men exakta guide för att ge dig omfatta
 400 nu sjuk i magsjuka
Senaste nyheterna om infektionssjukdomar I gamla tider hade även de rika parasiter CDC varnar för ökning av rabies kopplat till fladdermöss E. Coli-utbrott i 6 stater från förpackade sallader Listeri
400 nu sjuk i magsjuka
Senaste nyheterna om infektionssjukdomar I gamla tider hade även de rika parasiter CDC varnar för ökning av rabies kopplat till fladdermöss E. Coli-utbrott i 6 stater från förpackade sallader Listeri
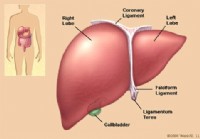 Levers anatomi och funktion
Leverdefinition och fakta Levern är ett stort, köttigt organ som sitter på höger sida av magen. Levern har två stora delar, som kallas höger och vänster lob. Levern är ett viktigt organ som har
Levers anatomi och funktion
Leverdefinition och fakta Levern är ett stort, köttigt organ som sitter på höger sida av magen. Levern har två stora delar, som kallas höger och vänster lob. Levern är ett viktigt organ som har