Abstract
Bakgrunn
Som en markør for Helicobacter pylori metodikk en entropi-basert beregning ble brukt til å påvise viktige rester av CagA mellomliggende sekvenser som magekreft biomarkør. For hver rest ble både kombinatorisk entropi og bakgrunn entropi beregnet, og entropi forskjell ble brukt som kriterium for funksjonen rest valg. De har verdiene ble deretter matet inn Support Vector Machines (SVM) med Radial Basis Function (RBF) kernel, og to parametre ble innstilt for å oppnå optimal F-verdien ved hjelp av rutenettet søk. To andre populære sekvens klassifiseringsmetoder, BLAST og HMMER, ble også brukt til samme data for sammenligning. Vår metode oppnådde 76% og 71% klassifisering nøyaktighet for vestlige og østasiatiske undergrupper, henholdsvis som utførte betydelig bedre enn BLAST og HMMER. Denne forskning viser at små variasjoner av aminosyrer i de viktige rester kan føre til virulens variansen av CagA stammer som resulterer i forskjellige gastroduodenal sykdommer. Denne studien gir ikke bare et nyttig verktøy for å forutsi sammenhengen mellom romanen CagA belastning og sykdommer, men også en generell nytt rammeverk for å avdekke biologiske sekvens biomarkører i befolkningsstudier Citation. Zhang C, Xu S, Xu D (2012) risikovurdering av magekreft forårsaket av Helicobacter pylori Redaktør: Niyaz Ahmed, University of Hyderabad, India mottatt: 13 november 2011; Godkjent: 11 april 2012; Publisert: May 15, 2012 | Copyright: © 2012 Zhang et al. Dette er en åpen-tilgang artikkelen distribueres under betingelsene i Creative Commons Attribution License, som tillater ubegrenset bruk, distribusjon og reproduksjon i ethvert medium, forutsatt den opprinnelige forfatteren og kilden krediteres Finansiering:. Dette arbeidet ble delvis støttet av amerikanske National Institute of Health [bevilgning antall R21 /R33 GM078601] og International Exchange og samarbeid Office of Nanjing Medical University, Kina. Finansiører hadde ingen rolle i studiedesign, datainnsamling og analyse, beslutning om å publisere, eller utarbeidelse av manuskriptet Konkurrerende interesser:.. Forfatterne har erklært at ingen konkurrerende interesser eksisterer Innledning Helicobacter pylori (H. pylori) Som en markør for H. pylori Innenfor den variable delen av CagA, er det noen forskjellige mellomliggende sekvenser mellom disse Epiya motiver. Ett eksemplar av Epiya pluss intervenerende sekvens blir identifisert som en Epiya segment. Fire unike typer Epiya segmenter har blitt funnet i CagA, definert som Epiya-A, -B, -C og -D [11]. Den CagA isolert fra østasiatiske land, utpekt som østasiatiske CagA, inneholder Epiya-A, Epiya-B og Epiya-D motiver. Den CagA fra vestlige land, Epiya-D, er erstattet av Epiya-C. Sterkere fosforylering motiv bindingsaktiviteten til Epiya-D-motivet fører til større morfologiske endringer enn hva Epiya-C motiv kan føre til i infiserte celler [11]. Det er denne Epiya-D motiv økte bindingsaktivitet og resulterende morfologiske endringer som identifiserer den som en mulig faktor for å forklare høyere forekomst av magekreft i østasiatiske land [23], [24]. Tidligere studier avdekket en variasjon i antall Epiya motiv gjentas for både Øst asiatiske og vestlige CagA, noe som kan påvirke biologiske aktiviteter. Yamaoka et al. [25] fant at i Columbia og USA, evne til CagA-positive H. pylori På grunn av den komplekse og variantsekvenser i CagA, relasjonene mellom polymorfisme av CagA og kliniske sykdommer bli en meget interessant problemstilling. Men de molekylære mekanismene som ligger bak ulike gastroduodenalsår sykdommer forårsaket av CagA-positive H. pylori I denne artikkelen foreslår vi en systematisk metode for å analysere ikke bare antall Epiya motiver i CagA sekvenser, men også bestemt rekkefølge mønstre av mellomliggende regioner. Først introduserer vi entropi beregning for å detektere de aminosyrerester i det variable området av CagA som gastrisk kreft biomarkører. Da vi ansette en overvåket læring prosedyre for å klassifisere kreft og ikke-kreft ved hjelp av informasjonen over oppdagede rester i CagA som funksjonene. Vi velger støttevektormaskiner (SVM) som en binær klassifikator og sammenligne vår metode med andre. Vår tilnærming beviser ikke bare vår hypotese at sekvensen av variabel region av CagA inneholder informasjon til å skille ulike sykdommer, men gir også et nyttig verktøy for å forutsi sammenhengen mellom romanen CagA belastninger og sykdommer og for å oppdage biomarkør også. Metoder data~~POS=TRUNC Forbehandling Basert på den tidligere beskrivelsen i Ref. [15], som er oppkalt vi Epiya motivet og de følgende mellomliggende regioner R1, R2, R3, R3 ', R4 og R4' (figur 1). Figur 2 viser posisjonen forholdet mellom Epiya motiv (R1) og andre mellomliggende områder ved hjelp av CagA typene A-B-D (Øst-Asia subtype) og A-B-C (vestlig undertype) som eksempler. R2 er relativt konservert på tvers av begge undertyper, men det er betydelige forskjeller mellom de mellomliggende områder R3 og R3 ', så vel som mellom R4 og R4'. The East Asian subtype og den vestlige subtype ble behandlet som to uavhengige grupper. Deres data ble deretter behandlet og resultatene ble analysert innenfor hver gruppe individuelt. Alle mellomliggende regioner ble hentet fra CagA sekvenser og sette inn de tilsvarende subtype grupper, og deretter flersekvenssammenstillinger ble brukt for hver gruppe individuelt ved hjelp Clustal X versjon 2.0.3 [29]. Sekvensene profilene (figur 1) ble bygget ved hjelp av Weblogo 3 [30]. Siden CagA er knyttet til nesten alle gastroduodenale sykdommer og enkel analyse av Epiya motiv gjentar ikke gi noen statistisk signifikante forskjeller mellom de sykdommene, den informasjon som indikerer en spesifikk sykdom kan være skjult i de mellomliggende områder. Denne forskning forutsetter at det er et sett av rester eller rester kombinasjoner som kan være nyttig som en markør for en bestemt sykdom. Denne studien fokuserer på magekreft og kreft anvender de /ikke-cancer grupper som eksempel. Basert på de fluktende sekvenser for hver mellomliggende region, ble spesifikke rester identifisert ved å sammenligne differansen av kombinatorisk entropi [31] mellom kreft og ikke-cancer grupper. Denne fremgangsmåten omfatter følgende trinn: Først av alt, vi dele de gitte flere justeringer for alle mellomliggende regioner i to grupper: magekreft gruppe og ikke-kreft gruppe. For hver kolonne med flere justeringer, beregner vi bakgrunnen entropi (. Eq 1) og kombinatorisk entropi (. Eq 2), beskrives som følger: (1) hvor representerer antall sekvenser i gruppe k Deretter entropien forskjellen mellom kombinatorisk entropi og bakgrunnen entropi beregnes:.. (3) Figur 3 viser entropi konseptet ved hjelp av tre ekstreme tilfeller. I tilfelle P1, aminosyrene er "tilfeldig og jevnt fordelt" over alle grupper og det er ingen vesentlig konservert mønster for denne stillingen. Sak P2 representerer en "globalt konservert 'mønster og alle aminosyrer er de samme i begge gruppene. I tilfelle P3 er noen spesifikke aminosyrer bare konservert i bestemte grupper, og ulike grupper har forskjellige aminosyrer. Vi kaller dette tilfellet 'lokalt bevart ". Ifølge beregningsresultatene av entropi forskjell for de ovennevnte tre tilfellene, er det kombi entropi for både' globalt bevart" og "lokalt bevart" tilfeller. For "tilfeldig og jevnt fordelt 'tilfelle får maksimal verdi. Vi kan skille de "bevart" og "tilfeldig og jevnt fordelt" tilfeller basert på kombi entropi, men det hjelper ikke plukke 'lokalt bevart' sak fra alle "konservert" tilfeller. Når vi ser på bakgrunn entropi på samme tid, blir den maksimale verdi, og 0-medium verdi for «tilfeldig og jevnt fordelt 'tilfelle' globalt konservert 'tilfelle' lokalt konservert 'tilfelle, henholdsvis. Endelig forskjellene for de ovennevnte tre tilfeller er :,, og får den minste verdi. Derfor er det entropi forskjellen en skikkelig måling for å oppdage en "lokalt konservert 'sekvens mønster. Basert på beregningene over, kan det bestemmes at riktig gruppering kan minimere entropien forskjellen for de rester som tilhører den "lokalt konservert 'tilfelle. For å utføre en test, blir en sekvens valgt mens resten av sekvensene er delt inn i en magekreft gruppe og en ikke-kreft gruppen. For alle valgte rester, blir den valgte sekvens plasseres i magekreft gruppen for å beregne forskjellen entropi, og da det er plassert i ikke-kreft gruppen for å få den tilsvarende entropi forskjell. Endelig er innhentet for alle valgte rester som blir brukt som funksjonen entropi. datasett. Vi søkte National Center for Biotechnology Information (NCBI ), fått Swiss-prot /bev og DDBJ protein database og 535 stammer av H. pylori Figur 4 viser arbeidsflyten av klassifiseringen /prediksjon prosedyre: bootstrapping. et stort problem i å bygge en klassifiseringsmodell i dette tilfellet er det stor forskjell på de utvalgsstørrelser mellom kreft og ikke-kreft grupper, som kan føre til skjevhet i klassifiseringsresultatene. En bootstrapping prosedyre ble brukt for å løse dette problemet. I hver subtype gruppe, for hvert opplærings /testdatasett, ble alle ikke-cancerprøver tatt, og deretter stammer ble kontinuerlig trukket fra kreft gruppe på en vilkårlig basis inntil nå den samme størrelse av de ikke-kreft gruppen. I dette tilfelle ble alle de tilgjengelige data som brukes selv om kreftprøvene ble anvendt flere ganger, gitt deres mindre størrelse i forhold til den ikke-kreft gruppen. Denne prosedyren ble anvendt fem ganger for å generere fem uavhengige treningssett for hver testsekvens. Klassifiseringen /prediksjon Resultatet er gjennomsnittet av de fem uavhengige resultater. Fordi datastørrelsen er liten, en leave-one-out (Loo) kryssvalidering prosedyre ble utført. Dette er ikke bare en vurdering av klassifikator ytelse på trening /testdata, men også et anslag på prediksjon kraft for nye tilfeller. Vi valgte SVM som binær klassifikator og brukt funksjons entropi vektorer for å trene og teste klassifikator. I tilfellet av to-klasse myk margin klassifisering, er beslutningen funksjon en vektet lineær kombinasjon definert som følger: (4) hvor representerer en brukerdefinert kjerne funksjon som måler likhetene mellom inngangsfunksjonen vektor og funksjonssvektorer i trenings datasett. er den vekt som er tilordnet trening funksjonen vektoren, og angir hvorvidt en CagA stamme er blitt merket med den positive klasse (1) eller negativ klasse (-1). Primal optimaliseringsproblem tar form: minimere (5) lagt (6) der. m er det totale antall stammer. er en slakk variabel som måler graden av feilklassifisering av det datum. er en kostnad parameter som åpner for handel av trening feil mot modellen kompleksitet. w er den normale vektor og b er utlignet. Når man sammenligner resultatene av polynomisk, tanh og Gaussiske radielle basis kjerner, vil resultatet oppnådd med RBF kjernen arbeidet best, hvor Gaussisk radielle basis kjerner (RBF :) er for generell læring når det ikke er noen forkunnskaper om dataene. Den SVM Lyspakke (http://svmlight.joachims.org/) [32] ble ansatt for å bygge vår søknad. Parametrene og var innstilt for å få den beste modellen for treningsdataene som er vist i det følgende. Alle andre SVM parametere ble satt til standardverdier Ytelse evaluering For å evaluere resultatene av klassifikator, er en rekke resultatmål brukt.. Nøyaktighet, sensitivitet og spesifisitet. En sann positiv (TP) er en kreft-relaterte sekvens klassifisert som sådan, mens en falsk positiv (FP) er en ikke-kreftrelatert sekvens klassifisert som kreft-relatert, en falsk negativ (FN) er en kreftrelatert sekvens klassifisert som ikke- -cancer relaterte og en sann negativ (TN) er en ikke-kreftrelatert sekvens klassifisert som ikke-kreft relatert. Nøyaktigheten, følsomhet (Sn), spesifisitet (Sp) og Matthews korrelasjonskoeffisient (MCC) for klassifisering er definert som følger: (7) (8) (9) (10) Siden det er bare to parametere for RBF kjernen og de er uavhengige, søkte vi et rutenett-søk for å finne de optimale parametrene for klassifikator. Vi brukte en harmonisk gjennomsnitt av sensitivitet og spesifisitet som målfunksjonen for å optimalisere ytelsen til modell for treningssettet, som er definert som følger: Resultater Residue Detection og Feature Beregnings Tabell 1 lister opp alle registrerte nøkkel rester ved å beregne entropien forskjellen i hver mellomliggende region for både vestlige og østasiatiske subtyper. Selv om det er noen geografiske variasjoner av CagA sekvenser mellom den vestlige og østasiatiske subtyper, kan noen vanlige rester fortsatt bli funnet å skille kreft og ikke-kreftgrupper. Det tyder på at disse rester kan være meget viktig ved bestemmelse virulensen av CagA og forholdet mellom CagA og noen spesifikke sykdommer. Residuet posisjonene er vist i figur 5. En tidligere studie [27] viser at forskjellige Epiya segmenter kan binde seg til de forskjellige kinaser, for eksempel, Epiya-R2 og Epiya-R3 /R3 'binder seg til den C-terminale Src kinase (CSK) mens Epiya-R4 og Epiya-R4' binder seg til SHP-2 kinase til å forårsake kolibrien fenotype. Den CagA-CSK interaksjon nedregulerer CagA-SHP-2 signal som perturbs cellulære funksjoner for å kontrollere virulens CagA. Det er funnet at de fleste detekterte residuer hører til R2 og R3 /R3 'områder og noen få rester i R4 /R4' regioner har blitt detektert. Dette kan være fordi R4 /R4 'har mer konservert sekvens enn R2, og R4 /R4' er kortere enn R3 /R3 '. Vi foreslår at de forskjellige rest mønstre i R2 eller R3 /R3 'regioner kan endre evne til å nedregulere CagA-SHP-to signalering, derfor endrer virulens CagA. Ren et al. fant at CagA multimerizes i pattedyrceller [33]. Dette multimerization er uavhengig av tyrosinfosforylering, men det er relatert til "FPLxRxxxVxDLSKVG" motiv som er oppkalt CM motiv i R3 'mellomliggende region. Siden multimerization er en forutsetning for den CagA-SHP-2 aliserte kompleks og etterfølgende deregulering av SHP-2, spiller CM motivet en viktig rolle i CagA-positiv H. pylori Mens de viktigste rester oppdaget kan avsløre noen forskjell mellom kreft og ikke-cancer grupper, kan ingen enkelt rest være en markør for kreft, som vist i figur 5. Denne forskning forutsier at en spesiell kombinasjon av alle eller delvis detekterte rester kan ha en høy korrelasjon med en spesiell sykdom. Hvis du vil kontrollere, flere lineære statistiske modeller, f.eks lineær regresjon og logistisk regresjon, ble påført på de detekterte funksjoner for å vurdere betydningen av hver rest og korrelasjonen mellom valgte rester og kreft. Ingen av modellene ovenfor, var i stand til å produsere et statistisk signifikant resultat. Siden funksjonene ikke kan monteres av enkle lineære modeller for å forutsi kreft, bruk en maskin læringsmetode for å analysere og klassifisere disse dataene blir nødvendig. Ved hjelp av den vestlige subtype gruppen som eksempelet, en løs grid-søket ble uroppført på og (figur 6A) og funnet ut at det beste er rundt for å få den høyeste F-verdien med loo kryssvalidering satsen 76%. Da et finere gitter søk ble gjennomført på området og en bedre F-verdi ble oppnådd med 79,7% LOO kryssvalidering ved. Samme prosedyre ble benyttet for østasiatiske subtype gruppe og best LOO kryssvalidering satsen 72,6% ble nådd. Siden det ikke er noen tidligere studier eller beregningsmetoder om samme emne, evaluere resultatene av denne forskning nye metoden er vanskelig. For å vurdere informasjonsinnholdet av sekvensene i form av deres kresne kraft for å forutsi kreft, ble en tilfeldig stokking fremgangsmåte som ble anvendt for å bygge kontrollgruppen. Først ble alle sekvenser fra den vestlige subtype plassert sammen for å bygge en sekvens basseng. For det andre, vi tilfeldig plukket samme antall sekvenser som kreft gruppe fra sekvensen bassenget og behandlet resten av sekvensene som de ikke-kreft gruppen. Deretter ble hele treningen prosedyren brukes til nylig stokket data for å finne den beste. De ovennevnte fremgangsmåten ble gjentatt fem ganger for å generere fem uavhengige stokkes datasett. Den med høyest F Det er i hovedsak tre kategorier av sekvens klassifisering metoder: funksjon basert, sekvens avstand basert og modellbasert. Metoden som vi beskrev i denne artikkelen tilhører den funksjonsbaserte kategorien. Vi valgte to av de mest populære sekvens klassifiseringsverktøy som representant metoder for andre to kategorier for sammenligning. BLAST [36] ble valgt for sekvensavstandsbasert kategori, siden det er den mest brukte sekvenssammenligning verktøyet. For modellbaserte kategorien, er den skjulte Markov modellen den typiske metoden for sekvensanalyse og dens mye brukt verktøy, HMMER [37], ble valgt. For klassifisering prosedyren for både BLAST og HMMER, vi brukte standardparameterne for verktøy, anvendt samme LOO kryssvalidering som vår metode, og brukt samme evaluerings formler oppført i Method delen. Tabell 2 viser klassifiserings resultater for alle tre metodene. Den SVM Fremgangsmåten fungerer betydelig bedre enn de to andre tilnærminger. BLAST oppnådd nær nøyaktigheten av entropi-SVM-metoden, men det forutsagte mange falske negativer med lav følsomhet. HAMMER oppnådd høy følsomhet, men med litt spesifisitet. Vurderer F Klassifiseringen resultat og konturen plottet (figur 6) sterkt støtte vår hypotese, dvs. informasjonen om de valgte rester i mellomliggende områder kan brukes til å klassifisere forholdet mellom CagA sekvenser og magekreft, selv om forskjellen mellom profilene av kreft og ikke-cancer gruppene er ikke meget sterk. Sammenligning mellom ulike sykdommer H. pylori Siden de fleste data ble samlet inn fra offentlige databaser uten nøyaktig diagnose informasjon før vi legger vår metode for å CagA data, vi kuratert sykdommen kommentarene manuelt for alle stammer av gjennomgang av litteratur. Tabell S1 viser fordelinger av alvorlige sykdommer for både vestlige og øst Asain subtype grupper. På grunn av den begrensning av strekk antall av noen sykdommer, så som atrofisk gastritt (AG) og magesår (GU), vi til slutt plukket kronisk gastritt (CG) og duodenal sår (DU) som kontrollgruppene for evaluering. DU gruppen i østasiatiske subtype inneholder 79 stammer, og en bootstrapping prosedyre ble brukt på alle andre grupper til å gjøre det samme antall stammer som østasiatiske DU gruppe. Dette trinnet sikrer alle sammenligninger på samme skala, siden verdien av kombinatorisk entropi er avhengig av antallet av sekvenser. Vi brukte formel (3) for å beregne differansen entropien av hver stilling mellom GC og CG /DU-grupper, og deretter summeres alle entropi forskjeller som den totale forskjellen mellom GC og CG /DU-grupper, som vist i Tabell S2. Ved å sammenligne resultatene mellom to grupper innenfor samme geografiske subtype (østasiatiske eller Western subtype), er det i samsvar med klinisk oppfatning at gastritt har sterkere relasjoner til kreft enn å DU [39] (generelt, gastritt tilfeller kan inneholde noen urapportert eller udiagnostisert kronisk atrofisk gastritt og intestinal metaplasi tilfeller med pasienter som har høy risiko for å utvikle GC). Ved å vurdere det samme sykdoms par mellom to geografiske undergrupper, det også forklart virulente forskjellen mellom øst asiatiske og vestlige subtyper. I tillegg, på grunn av den høye likheten mellom ulike sykdomsgrupper av østasiatiske subtype, selv med mer data, har vi fortsatt ikke kan nå samme klassifisering nøyaktighet som den vestlige subtype gruppen. Basert på resultatene ovenfor CagA sekvenser viser potensial for å skille flere gastroduodenalsår sykdommer. For å evaluere klassifisering resultatene har vi DU gruppe for å erstatte ikke-kreft gruppen, og deretter påført hele klassifiseringen prosedyren igjen uten bootstrapping, siden disse to sykdommer gruppene har sammenlignbare størrelser. Tabell S3 viser klassifiseringsresultatene. Selv om fra klinisk synspunkt, har DU den negtive sammenheng med GC blant alle gastroduodenalsår sykdommer [40], ble klassifiseringen resultatene av to subtype grupper bare litt bedre. Dermed kreftrelaterte CagA stammer kan ha noen unike sekvens mønstre sammenligne med alle andre gastroduodenalsår sykdommer. Derfor innstiller en undergruppe av kontrollgruppen kan ikke være i stand til å forbedre klassifiseringen nøyaktighet. Selv om forskning viser at det er sekvensen markører for å skille mellom kreft gruppe og ikke-kreft gruppe de store profilene i disse to gruppene er for like til å skille ved hjelp av tradisjonelle metoder siden CagA sekvenser er samlet sterkt konservert. Derfor har vi fokusert på å identifisere de informative rester, kvantifisere informasjon om disse utvalgte rester, og deretter bruke den til å lage en klassifikator som kan forutsi om en ny sekvens tilhører kreft gruppen eller ikke-kreft gruppe. Denne metoden ikke bare belyser forholdet mellom CagA sekvenser og magekreft, men også kan gi et nyttig verktøy for magekreft diagnose eller prognose. Mekanismene for H. pylori
, cytotoxin-assosiert genet A (CagA) har blitt avslørt å være den viktigste virulens faktor som forårsaker gastroduodenalsår sykdommer. Imidlertid, de molekylære mekanismer som ligger til grunn for utviklingen av forskjellige gastroduodenal sykdommer forårsaket av CagA-positive H. pylori
infeksjon er fortsatt ukjent. Aktuelle studier er begrenset til evalueringen av sammenhengen mellom sykdom og antallet Glu-Pro-Ile-Tyr-Ala (Epiya) motivene i CagA belastning. For ytterligere å forstå forholdet mellom CagA sekvens og dens virulens til magekreft, foreslo vi en systematisk entropi basert tilnærming for å identifisere kreftrelaterte rester i de mellomliggende regionene CagA og ansatt en overvåket maskinlæringsmetode for kreft og ikke-krefttilfeller klassifisering.
Konklusjon
Bruke CagA Sequence Markers. PLoS ONE 7 (5): e36844. doi: 10,1371 /journal.pone.0036844
er en Gram-negative helix formet bakterie som lever på den menneskelige magen og infisere mer enn halvparten av verdens befolkning [1], [2], [ ,,,0],3]. Nyere studier har vist at det er forbundet med gastroduodenale sykdommer, inkludert duodenalsår [4], magesår [5] og kronisk gastritt. Enda viktigere er det en betydelig risikofaktor for utvikling av magekreft [6], [7], [8]. Det har blitt klassifisert som et klasse 1 menneskelige kreftfremkallende av Verdens helseorganisasjon siden 1994 [1].
, den cytotoksin-assosierte gen A (CagA) har blitt avslørt ved ytterligere analyse for å være den viktigste virulens faktor. H. pylori
stammer som bærer den CagA genet øke risikoen faktor på gastroduodenale sykdommer ved å tre glende CagA-negative stammer [6], [9], [10]. CagA, som er kodet av CagA genet, er en 125 til 140 kDa protein. Den inneholder 1142-1320 aminosyrer og har en variabel region ved den C-terminale region hvori forskjellige korte sekvenser (for eksempel Epiya motiv) gjentas 1-7 ganger. Etter H. pylori
kolon på overflaten av den gastriske epitel, kan CagA blir translokert inn i epiteliale gastrisk cellen gjennom en type IV sekresjon system. Når injisert inn i vertscellen, CagA lokaliserer til plasmamembranen, og kan bli fosforylert av Src-familie-tyrosin-kinaser på spesifikke tyrosinrester av en fem-amino-syre (Epiya) motivet [11], [12], [13] , [14]. Tyrosin-fosforylert CagA binder så spesifikt til SHP-2 tyrosin fosfatase 11,15 for å aktivere en fosforylase, noe som fører til en kaskade effekt som forstyrrer signaltransduksjonsbane av vertscellen, som fører til en restrukturering av vertscellen cytoskjelettet og dannelse av hummingbird fenotype [11], [16]. På samme tid gjennom aktivering av mitogen-aktivert proteinkinase (MAPK), ekstracellulære signalregulerte kinase (ERK) [17] og fokal adhesjonskinase (FAK), CagA også kan føre til celle dissosiasjon og infiltrerende tumorvekst [18], [19 ], [20], [21]. En slik prosess gjør CagA en viktigste virulens faktor i H. pylori product: [22].
å forårsake gastrisk mucosal atrofi og intestinal metaplasi kan være relatert til antall Epiya motiver i CagA belastning. Argent et al. [16] kom til samme konklusjon senere. Imidlertid ble i motsetning meninger publisert av Lai et al. [26] basert på funn av ingen sammenheng mellom antall Epiya motiver i CagA belastning og klinisk sykdom innen 58 isolater fra Taiwan. Med tanke på størrelse og geografisk begrensning av disse studiene, gyldigheten av denne konklusjonen er tvilsom. Bortsett fra antallet Epiya motiv gjentar sekvensen differansen av stammer i variable områder også kan føre til en betydelig forskjell av virulens, som kan relateres til forskjellige sykdomsfremkallende egenskaper på H. pylori product: [27].
infeksjon er fortsatt ukjent. Inntil nå har de fleste studier er fortsatt begrenset til oppdagelsen eller evaluering av sammenhengen mellom antall CagA Epiya motiver og sykdommer [28].
rester gjenkjenning
. indikerer antall rester av typen i kolonnen i
av gruppen k
. er antall rester av typen i kolonnen i
. representerer det totale antall sekvenser i innretting (2) hvor
Feature-entropi Beregning
Klassifisering av CagA Sekvenser
CagA protein. Blant dem er det 287 østasiatiske subtype stammer og 248 vestlige subtype stammer. I østasiatiske subtype gruppe, 47 ut av 287 stammer er fra mage kreftpasienter og resten er fra andre sykdommer. I den vestlige subtype gruppe, det er 37 stammer fra mage kreftpasienter, og restene er fra andre sykdommer eller normale kontroller, inkludert 24 stammer fra frivillige som helse (sykdom) status var ukjent.
Arbeidsflyt.
Cross-validering.
SVM.
(11)
formidlet mage patogenesen. Med flere CM motiver H. pylori
stammer er mye sannsynlig assosiert med alvorlige gastroduodenalsår sykdommer [33], [34], men denne observasjonen kan ikke forklare hvorfor ulike gastroduodenalsår sykdommer kan bygges ut med nøyaktig samme antall CM motiver. Vår undersøkelse påvist to rester i CM motiv av R3 'mellomliggende region, som kan føre til forandring av multimerization, dermed endre virulens CagA. Dette er i samsvar med tidligere funn [35] at sekvensen forskjellen mellom østasiatiske CM og den vestlige CM bestemmer bindende affinitet mellom CagA og SHP-2.
Parameter Training for klassifisering
verdi, noe som tilsvarer 46,6% ble valgt og dens kontur plott er vist i figur 6B. Dette tilfeldig stokking Evalueringen ble også brukt til østasiatiske subtype data og den beste F
verdien var på 54,3%. Sammenligning av to tomter viser signifikant forskjell på F
verdier mellom data med riktig gruppering av kreft og ikke-krefttilfeller i opplæring og de beste tilfeldig stokket data. Resultatet tyder på at de mellomliggende regionene er informative å skille mellom kreft og ikke-kreft grupper og vår metode kan bruke informasjonen effektivt.
Klassifisering ytelse
verdier og MCC
verdier, prediksjonsresultater fra BLAST og HAMMER er nesten tilfeldig.
infeksjon er assosiert med de fleste gastroduodenalsår sykdommer, blant annet magekreft er den mest alvorlige en forårsaker mer enn 700.000 dødsfall på verdensbasis hvert år [38]. Siden H. pylori
er en viktig risikofaktor for magekreft (GC), oppdagelsen av mekanismen for H. pylori
formidling GC blir en topp prioritert oppgave i dette feltet. Sammenlignet med andre sykdommer, diagnostisering informasjon om GC fra offentlige data er relativt nøyaktig, og det er en annen viktig grunn til å fokusere på GC i dette papiret. Våre studier er ikke begrenset til GC, skjønt. Vi prøvde også å vurdere forholdet mellom variansen CagA sekvenser og ulike sykdommer.
Diskusjoner
forårsaker de forskjellige gastroduodenal sykdommer er fortsatt uklart, men det er sannsynlig at forskjellige gastroduodenal sykdommer forårsaket av H. pylori
infeksjon aksje noen sekvens mønstre i de mellomliggende regioner. Små variasjoner av aminosyrer i de viktige rester kan føre til virulens variansen av CagA stammer som resulterer i forskjellige gastroduodenal sykdommer. Mens CagA kan være en markør for å oppdage potensielle kreftrisiko ved bruk CagA alene å skille alle gastroduodenalsår sykdommer er ikke realistisk. Som en fremtidig studie, vil vi utvikle nye modeller som skiller ulike gastroduodenalsår sykdommer fra CagA og andre gener.
Hjelpemiddel Informasjon
Tabell S1. .
Antall stammer i hver sykdom
doi: 10,1371 /journal.pone.0036844.s001 plakater (DOC)
Tabell S2. .
Total entropi forskjell mellom magekreft og to andre sykdommer grupper
doi: 10,1371 /journal.pone.0036844.s002 plakater (DOC)
tabell S3.
Klassifisering ytelse mellom magekreft og duodenalsår grupper for både den vestlige og østasiatiske subtyper
doi:. 10,1371 /journal.pone.0036844.s003 plakater (DOC)
 Hvis du er over 50,
Hvis du er over 50,
 Diagnose av virusinfeksjoner ved hjelp av mikro- og nanoskala -teknologi
Diagnose av virusinfeksjoner ved hjelp av mikro- og nanoskala -teknologi
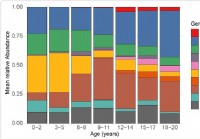 Endring av mikrobiom i øvre luftveier hos barn relatert til følsomhet for SARS-CoV-2
Endring av mikrobiom i øvre luftveier hos barn relatert til følsomhet for SARS-CoV-2
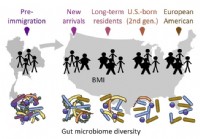 Migrasjon påvirker tarmmikrobiota som igjen påvirker helsefunnforskere
Migrasjon påvirker tarmmikrobiota som igjen påvirker helsefunnforskere
 RNA -sekvensering gir ny innsikt i mikrobiomet
RNA -sekvensering gir ny innsikt i mikrobiomet
 Tarmmikrober i veps hjelper til med å overvinne plantevernmidler
Tarmmikrober i veps hjelper til med å overvinne plantevernmidler
 Antioksidanter i kostholdet kan øke risikoen for tarmkreft,
ny studie avslører Helsemessige fordeler av antioksidanter i mat har blitt bevist gjennom en betydelig mengde vitenskapelig litteratur. Nå, en ny studie viser at for mye av det gode kanskje ikke er så
Antioksidanter i kostholdet kan øke risikoen for tarmkreft,
ny studie avslører Helsemessige fordeler av antioksidanter i mat har blitt bevist gjennom en betydelig mengde vitenskapelig litteratur. Nå, en ny studie viser at for mye av det gode kanskje ikke er så
 Crohns sykdom
Crohns sykdom forårsaker betennelse i mage -tarmkanalen. Det kan forveksles med ulcerøs kolitt og irritabel tarm, men Crohns sykdom er unik for seg selv. På Ogden Clinic GI på McKay, vi kan diagnostis
Crohns sykdom
Crohns sykdom forårsaker betennelse i mage -tarmkanalen. Det kan forveksles med ulcerøs kolitt og irritabel tarm, men Crohns sykdom er unik for seg selv. På Ogden Clinic GI på McKay, vi kan diagnostis
 Det du spiser kan endre måten antibiotika påvirker tarmen på
En ny studie av forskere ved Brown University i Rhode Island har funnet ut at diett kan påvirke hvordan tarmmikrobiomet påvirkes av behandling med antibiotika. Forskerne undersøkte hvordan antibiotika
Det du spiser kan endre måten antibiotika påvirker tarmen på
En ny studie av forskere ved Brown University i Rhode Island har funnet ut at diett kan påvirke hvordan tarmmikrobiomet påvirkes av behandling med antibiotika. Forskerne undersøkte hvordan antibiotika