Identifikácia DNA zmenami metylácie spojená s ľudskou rakovinou žalúdka
abstraktné
pozadia
epigenetické zmeny génovej expresie je bežná udalosť v ľudskej rakoviny. Metylácie DNA je známy epigenetické proces, ale overenie presnú povahu epigenetických zmien spojených s rakovinou zostáva obtiažna.
Metódy
sme profilované na methylome ľudského žalúdka rakoviny tkaniva pri rozlíšení 50 bp s použitím methylata obohatenia DNA technika (methylata CPG puncový zotavenie ostrov) v kombinácii s analyzátorom genómu a nové normalizačného algoritmu.
Výsledky
sme boli schopní získať komplexný pohľad na promotérov s rôznymi CPG hustotách, vrátane CPG ostrovy (CGI), prepis orgány a rôzne opakovania triedy. Zistili sme, že rakovina žalúdka bola spojená s hypermetylace 5 'CGI a 5'-konca kódujúcich exónov, rovnako ako hypometylácii opakujúcich sa prvkov, ako sú krátke rozptýlených jadrových prvkov a kompozitného prvku SVS. Hypermetylace z 5 'CGI významne koreluje s down-reguláciu súvisiacich génov, ako sú napríklad tie, ktoré v HOx stroje a Histon génové rodiny. Zistili sme tiež dlhého doletu epigenetické umlčanie (LRES) regióny žalúdočné rakoviny tkaniva a identifikovaných viacero génov (hypermethylated MDM2
DYRK2 stroje a lyž
) v rámci týchto regiónov. Stav metylácie CGI a gén anotácie prvkov v metastatických lymfatických uzlinách bol prechodný medzi normálnou a rakovinové tkanivá, čo naznačuje, že metylácie špecifických génov je v rakovinové tkanive postupne zvyšuje.
Závery
našich zistení poskytne cenné údaje pre budúce analýzu CPG metylácie vzorov, užitočné markery pre diagnostiku rakoviny žalúdka, ako aj nové metódy pre analýzu pre vyšetrovanie klinických epigenomics.
pozadí
rakovina žalúdka je druhou najčastejšou príčinou úmrtí na rakovinu na celom svete po rakovine pľúc, čo vedie viac ako 800.000 úmrtí na celom svete ročne [1]. Aktuálne 5-ročné prežitie osôb s diagnózou karcinómu žalúdka je len 20 až 30%, s tým, že nízka miera súvisí s tým, že vo väčšine prípadov sú už v pokročilom štádiu, keď sú diagnostikované. Rovnako ako vo všetkých rakovín, včasná detekcia zostane najviac sľubný prístup pre zlepšenie prežitie. Preto, pochopenie príčin vzniku nádorov v ľudskej žalúdočnej tkanive je zásadný.
Infekcia H. pylori
je dobre zavedenou a bežnou príčinou rakoviny žalúdka. Avšak, zmeny v rôznych genetických faktorov sú tiež dôležité pri zvyšovaní rizika rakoviny žalúdka. Je dobre známe, že chromozomálne nestabilita pochádzajúca z genetických faktorov, ako je mikrosatelitních nestabilita, rovnako ako KRAS stroje a p53 mutácií
viesť k vývoju nádorov. Niekoľko genomické štúdie identifikovali zárodočnej mutácie v špecifických génov [2-4] a ochorenia citlivého loci [5, 6] pre rakovinu žalúdka. Nedávne štúdie porovnávajúce žalúdočné rakovinu a normálne tkanivo identifikovali niekoľko genetických markerov, vrátane diagnostických markerov [NF2
[7], INHBA
[8], SFRP4
[9]], prognostické markery [CD9
[10], CDH17
[11], PDCD6
[12]] a rakovinou žalúdka spojené s génmi [MUC13
[9], CLDN1
[13], Ki67
a CD34
[14]]. Okrem toho sa zistilo, že epigenetické mechanizmy, ako je metylácie DNA a histónov modifikácií, že sú dôležité pri regulácii expresie génov podieľajúcich sa na biológiu a ochorenia gastrointestinálneho traktu [15].
Metylácia DNA hrá zásadnú úlohu v eukaryoty a je spojená s mnohými kľúčových mechanizmov, vrátane imprinting, X chromozómu inaktiváciu, starnutie a karcinogenézy. Zmena metylácie DNA v genóme sa vyskytuje takmer vo všetkých typov rakoviny a môže viesť k zmenám v génovej expresii, ako je napríklad nadmerná expresia onkogénov a umlčanie nádorových supresorových génov počas vývoja rakoviny [16]. Niekoľko štúdií ukázalo, že akumulácia genetických a epigenetických zmien v žalúdku prekanceróznych lézií, môže mať vplyv na veľký počet cieľov, ako sú napríklad komponenty opravy DNA systém, nádorové supresormi, onkogény, regulátory bunkového cyklu, rastové faktory, a adhéznych molekúl [17-20] , Avšak, tieto štúdie boli zamerané predovšetkým na niekoľko kandidátnych génov alebo pokryté len časť celého genómu. Preto prístup k celkový prehľad o epigenetických zmien súvisiacich s vývojom rakoviny bolo ťažké. Jedná sa najmä o porozumení zmeny metylácie DNA v intragenic regiónoch, CPG ostrovov, intergenových regiónov a opakujte sekvencií zostáva obmedzená. V dôsledku toho je veľký záujem v genóme-široký analýzy aberantne metylácie DNA v týchto regiónoch. Apartmán V komplexného genómu meradle profilovania metylácie DNA v embryogenézy a karcinogenézy, s vysokým rozlíšením celých metódy sekvenovania genómu, ako je BS-nasl [21 -24], MEDIPO-seq [25, 26], a MethylCap-nasl [27-29] boli vyvinuté. Napriek rýchly vývoj sekvenovania založené na mapovanie techniky, stále existuje nedostatok komparatívneho výskumu, ktoré sú kritické pre klinické epigenomics štúdií, vrátane tých, zamerané na rakovinu. Na rozdiel od prístupov založených na microarray, sekvenačnej dáta sú vyrábané vo formáte, ktorý nie je prístupný diferenciálnej analýza a analýza pracovný postup nie je štandardizovaný. Z tohto dôvodu je potrebné výpočtovo nenákladné metódy normalizácie zvládnuť výpočtovej záťaže spracovanie veľkých rozmerov s vysokým rozlíšením sekvenčné dáta.
Tu sme zaviedli algoritmus normalizácie, ktorá berie do úvahy vzorke špecifické celkovú hustotu čítanie, priestorové distribúcie CPG loci a pozadia sekvenčného zaujatosti. Potom sme vytvorili ucelený celok-genómu methylome normálne žalúdočné tkaniva, rakovina žalúdka tkanivo a metastatických lymfatických uzlín pomocou MethylCap-nasl metódu a získať podrobné informácie o svojom rozrušenie počas karcinogenézy a metastáz. To je ľahko použiteľný pre porovnávacie analýzy methylomes a iných typov epigenomic dát, a to má určité dôsledky pre klinické epigenomics.
Metódy
vzoriek žalúdočnej tkaniva
Získali sme tri snap-mrazené nádorov žalúdka a uzavreté normálne žalúdka tkanivo od Seoul National University College of Medicine pre methylome štúdii. Okrem toho, bolo získané dvadsať osem spárovaných normálnych a nádorových tkanivách žalúdka pre ďalšie potvrdenie. Všetky vzorky boli získané endoskopickou resekciou pri vyšetrení pacientov, ktorí dali informovaný súhlas
metylovaných test pre obnovu DNA (MIRA)
genómovej DNA z 25 mg žalúdočné tkaniva sa čistí pomocou DNeasy Blood &.; Tissue Kit (Qiagen, Valencia, CA). Genomickej DNA vzorky od 3 jedincov boli zhromaždené v rovnakej koncentrácii. Mira bola vykonaná ako bolo opísané skôr [30 až 32]. Stručne povedané, GST značeného MBD2b a His-MBD3L1 proteíny boli pripravené, ako je popísané. 15 ug genómovej DNA bol roztrieštený na 100 ~ 500 bp sonikací a inkubované s 28 ug purifikovaného GST-MBD2b proteínu, 28 ug Jeho-MBD3L1 proteínu a 7 ug JM110 bakteriálnej DNA po dobu 6 hodín. Pridá sa 30 ul guľôčok MagneGST (Promega, Madison, WI) preblocked sa 7 ug JM110 bakteriálnej RNA a inkubujú sa pri teplote 4 ° C s otočným po dobu 45 minút v konečnom 600 ul Mira väzobných reakčnej zmesi. Perličky boli premyté trikrát 1 ml premývacieho pufra, a methylata fragmenty boli eluované inkubáciou pri teplote miestnosti po dobu 5 minút a potom 56 ° C počas 30 minút s 30 ul TE, obsahujúceho RNase A (100 ug, Qiagen) a na proteináza K ( 15 ug, Qiagen). Eluovanej DNA fragmenty boli ďalej čistené pomocou Qiaquick PCR purifikačnej súpravy (Qiagen).
Illumina Genome Analyzer sekvenčné
Použili sme 10 ng eluovanej DNA pre Illumina Genome Analyzer sekvenovania. Po podviazanie z dvojice SOLEX adaptérov, ligačních produktov s maximálnou veľkosťou inzertu 200 bp boli prečistené na gélu na 2% agarosovém a podrobená PCR amplifikáciu. Cluster generácie a 36 cyklov sekvenovania sa vykonávali podľa pokynov výrobcu. sekvenovania sme 120 ul adaptéra ligu, veľkostne frakcionované DNA (2 ~ 4 hodín) na Illumina Genome Analyzer. Sekvenčné značky boli mapované do ľudského genómu (hg18 databázu UCSC založené na NCBI Zostavte 36,1 zostavy) pomocou SOLEX Analysis Pipeline (verzia 0.3.0). Sequenced číta o 34 bp (s výnimkou prvého a posledného nukleotidu), ktorý prešiel boli použité filtre kontroly kvality. Spracovanie
dát a výpočet MES
Rozšírili sme 3'konci o 34 bp číta o 200 bp na pokrytie DNA fragmenty viazané proteíny LMD. Odpočet bol premenený na dátové súbory prehliadač rozšíriteľných (lôžko) pre vizualizáciu v UCSC genóm prehliadača http: //. Genóm UCSC edu /.. Počítali sme prekrývajúcich sa sekvenčných tagov v rozlíšení 50 bp. Nájsť obohatené genómovej regióny, počet mapované číta v posuvné okno 1 kb bol v porovnaní s celkovým počtom číta alebo počet pozadia čítanie v genóme. Ako taký, MES bola vypočítaná dvoma spôsobmi; jeden je ako log2 (cieľová čítanie počty /cieľovej veľkosti) /(celkový počet čítanie /veľkosť genómu) a podlahou na nulu, druhá je ako log2 (cieľová čítanie počty /cieľovej veľkosti) /(pozadie čítanie počty /pozadia veľkosť) a podlahou na nulu. Ak chcete nastaviť na pozadí sekvenovania zaujatosť, MESbg bola vypočítaná rovnakým spôsobom pre vstupné sekvenčné bez afinity čistenia a odpočíta sa od MES.
Genómovej pozície CGI, promotérov, prepis orgánov, CDS, a opakujúce sa prvky
Všetky genómovej pozície CGI, prepisy a opakujte prvky boli stiahnuté z UCSC genómu prehliadači. Celkom 27,639 CGI (s výnimkou náhodne nachádza CGI) bol predpovedal podľa nasledujúcich kritérií: GC obsah 50% alebo viac, dĺžka viac ako 200 bp, a pomer vyšší ako 0,6 pozorovaný počet CPG dinukleotid k predpokladanému počtu [33] , Referenčná sekvencia kolekcia NCBI mRNA (RefSeq od vydania verzie 46; March 11, 2011) bol použitý pre identifikáciu transkripčný jednotky s definovanou začiatku transkripcie, koniec stránky a CDS začiatok, koniec stránky. Pre predkladateľa sme použili oblasť 500 bp upstream ~ 500 bp po smere od počiatku transkripcie. Získali sme ~ 5 miliónov opakované lokality, ktoré boli stanovené podľa RepeatMasker program založený na knižnici RepBase opakovanie.
Úroveň metylácie genómovej prvkov
metylácie úrovni CGI, propagátor, génového tela, a opakujte prvok bola odhadnutá pomocou MES prekrývajúcich sa každý prvok. MES = 0 sa používa pre definovanie nemethylované prvky. Na meranie hypermetylace alebo hypometylácii na rakovinu, sme vypočítali diferenciálnej neporiadok ako (Cancer MES - Normal MES). Diferenciál MES > 1.0 bola použitá ako medzná hodnota. Pre pochopenie funkcie vybraných génov sme použili klasifikáciu ontológia génov cez DAVID Funkčné Anotácia Clustering nástroje http: .... //David ABCC ncifcrf gov /
analýza génovej expresie
microarray produkt používaný v tejto štúdii bola Codelink Human Genome Celá 55 K čip (GE Healthcare, USA). Všetky experimentálne postupy vrátane Crna cieľovej prípravy, hybridizácia, post-hybridizácia farbivo spojka boli vykonané za použitia dodávateľa odporúča protokolov. Súbory výsledkov boli importované do GeneSpring GX 7.3 (Agilent Technologies, USA) pre filtrovanie a základné štatistické analýzy. Medzi 55 K génov na čipe, len s génmi prítomných príznakov v aspoň 50% vzoriek boli vybrané pre následnú analýzu. Údaje microarray boli uložené na GEO http: .. //Www NCBI NLM NIH gov /GEO /(prístupové číslo GSE33651)
Mira a real-time qPCR
Mira ... bola vykonaná na štyroch ďalších jednotlivých vzoriek. DNA bola čistená zo supernatantu a monitorovaná v reálnom čase za použitia Roche qPCR 480 stroja. Sekvencie použité priméry sú uvedené v ďalšej súbor 1: Tabuľka č. S1
bisulfitového, metylácie-špecifické PCR a Pyrosequencing
sme izolované genomickej DNA z jednotlivého vzorky pomocou Qiagen DNeasy Tissue Kit (Qiagen). Bisulfitového bola vykonaná za použitia metylácie zlaté súpravy EZ DNA (Zymo Research) podľa inštrukcií výrobcu. Hydrogensiričitanovú upravené DNA uskladnili pri -80 ° C až do ďalšieho použitia. Primery použité pre MSP boli navrhnuté s použitím Methprimer [34], a sú uvedené v ďalšej súbor 1: Tabuľka S1. PCR bola vykonaná s HotStarTaq polymerázy (Qiagen) a zahŕňal počiatočné inkubácii pri 95 ° C počas 15 minút, nasleduje 40 cyklov 95 ° C po dobu 1 minúty, 59 ° C po dobu 1 minúty a 72 ° C počas 40 sekúnd, nasleduje jeden cyklus 72 ° C počas 10 minút. Produkty MSP boli oddelené na 2% agarosovém gélu a vizualizované farbením EtBr. Pyrosekvenační reakcie boli vykonané automaticky systémom PSQ 96 (Pyrosequencing AB) podľa inštrukcií výrobcu. Stručne povedané, biotinylizované produkt PCR (50 ul) sa čistí pomocou streptavidín-Sepharose guľôčky (Amersham Biosciences). Purifikovaný produkt bol vložený do reagenčnej kazety s enzýmu, substrátu a dNTP súčasťou stavebnice PSQ96 SNP Reagent (Pyrosequencing AB). Tieto sekvenčné priméry pre Pyrosequencing sú uvedené v ďalšej súbor 1: Tabuľka č. S1
Výsledky
spracovanie MIRA-nasl methylome údaje
čistí sme methylata DNA obohacuje MIRA (methyluje CPG testu zotavenie ostrov) a sekvenované DNA sekvenovania s použitím novej generácie. úrovne metylácie DNA boli stanovené za použitia sekvenčné čítanie počty zodpovedajúcich oblastí, v 50 bp intervaloch, ako je popísané v metódach. Vytvorili sme DNA metylácie mapy pre normálnu aj rakovinových žalúdočné tkaniva. Pre každú vzorku, sme získali asi 10 miliónov sekvencie číta (dodatočný súbor 1: Tabuľka S2). Každá methylome obsahovala ~ 140 miliónov CPG číta, pokrývajúca ~ 48% všetkých genómových miest CPG s výnimkou Centromera (ďalší súbor 1: Tabuľka S3). Priemerná pokrytie CPG číta v každej methylome bola 4.5x. Na podporu vysokej citlivosti Mira, genomické segmenty, ktoré obsahujú iba jednu CPG mal vyšší čítať počty než tí s žiadnym CPG (p
hodnota = 0), čo naznačuje, že jednotlivé zmeny CPG mohla byť vyriešená pomocou Mira. Priemerná sekvencie číta vzrástol v pomere k počtu CPG v 50 bp intervale, a v skutočnosti, MIRA pokrytie nebola nízka, a to aj pre regióny s nízkou hustotou CPG (ďalší súbor 2: Obrázok S1). Dohromady tieto výsledky ukazujú, že Mira bola úspešná pri odstraňovaní dostatočnú zlomok metylovaných oblastí. Čo sa týka presnosti Mira, ~ 99% MIRA-zachytených úlomkov malo aspoň jedno miesto CPG v rámci ich poradie, čo ukazuje na nízky počet falošných detekcie.
Pre meranie obohatenie miestnych metylácie signálov, sme vypočítali metylácie obohacovania uránu skóre (MES ) získaním prečítanú počet v danom regióne, a potom prevedením normalizáciu pre riadenie pre celkovú čítanie počtu (MEST) v ukážkovom (globálne normalizácia) alebo sa počet miestnych čítanie (MESl) v oblasti užívateľom definovanej obklopujúcu (miestne normalizácia) (pozri metódy). To umožňuje priame porovnanie nezávislých vzoriek s rôznou hustotou čítania. Potom sme vykonali logaritmickej transformácii odvodené skóre. Spolu s tým ďalšie matematické prednosti, toto poskytuje výhodu stabilizácie rozptylu, a to najmä pre vysoko na čítanie počty, ktoré sú často v spojení s vysokými technickými varianty, ktoré môžu zaviesť výrazné skreslenie v dátach.
Hodnotili sme štatistickú významnosť MES v dvoma spôsobmi. Randomizovanej MES boli generované numericky permutácií genomické pozície z našich sekvencie číta. Na pozadí MES (MESbg) bol experimentálne získaný sekvencovanie normálne genóm bez afinitná čistenia. Ako sa dalo očakávať, že reálne dáta priniesla výrazne vyššia obohacovania skóre (dodatočný súbor 2: Obrázok S2). Pozoruhodne, MESbg bola vyššia ako MES z randomizovanej genómov údaj, že na pozadí sekvencie sám môže vytvoriť obohatenie, pravdepodobne v dôsledku chromatínu dostupnosti a amplifikácie zaujatosti. V súlade s nedávnymi správami [35], to poukazuje na potrebu riadnej kalibráciu inherentnú sekvenovania zaujatosť. Preto sme normalizovať naše MES s MESbg.
Ak chcete nájsť optimálne podmienky pre normalizáciu, sme porovnávali štatistické vhodnosti rôznych metód normalizácie. Distribúcia značka pozdĺž genómu možno modelovať Poissonova rozdelenia [36, 37]. Dobrota fit bola testovaná pomocou testu Kolmogorov-Smirnov. V tomto teste, nízke D štatistika naznačuje dobrou voľbou. Kým model Poisson prekonal celkovo Gaussova, MES ukázal lepšiu kondíciu ako surové prečítaných počtov (prídavné súboru 2: Obrázok S3), ilustrujúci vzácna udalosť povahu čítať počtu opatrení log-zmenšený. Normalizované MESl kalibrovaná riadiacim sekvenovania (MESbg) poskytla ešte lepšie výsledky ako normalizovanú Mest kalibrované riadiacim sekvencovania (MESbg).
Global a chromozomálne výhľadom na DNA metylácie
po potvrdení najlepší spôsob bodovania úrovne metylácie genómu-široký na 50-bP intervaloch, sme sa prvýkrát skúmali chromozomálne metylačnej vzory normálnych vzoriek. Priemerné MES vypočítané pre každého chromozómu navrhnuté, že CPG-bohaté a gén bohatý na chromozómy majú tendenciu byť vysoko methylata (obrázok 1A). Hladiny metylácie chromozómov s veľkým množstvom dlhej rozptýlené jadrovej elementy (linky) boli relatívne nízke (napr., Chromozóm 4). Zaujímavé je, že množstvo krátkych rozptýlených jadrových prvkov (siny) bol úmerný chromozómu metylácie vzoru. To je pravdepodobne spôsobené tým, že sú zvyčajne sínusov zoskupený v oblastiach génu bohatej. Sexchromozómy boli globálne hypomethylated s nižšou hustotou CPG a vyšším obsahom opakované než autosomes. Vzhľadom k tomu, sme použili tkanív odobratých z muža v tomto experimente, globálne hypometylácii chromozómu X pozorované nie je spojený s X inaktiváciu. pohľady chromozómové celej zrekapituloval vysokú hustotu CPG a vysoká metylácie okolo génovo bohaté (pozri čierne pruhy v spodnej časti) a CGI bohatých regiónoch (pozri modré pruhy hore) (obrázok 1B). Na rozdiel od nízkej hustote CPG a nízke metylácie boli pozorované okolo krajov gény chudobných, ktoré boli bohaté na ďalekonosných opakovanie (viac ako 1 kb) (viď červené pruhy hore). Priemerné MES vyplýva, že úroveň metylácie CGI je podstatne vyššia ako u Génový oblastí alebo opakovanie (Obrázok 1B). Obrázok 1 metylačnej vzory normálny žalúdočné tkaniva. (A) chromozómov celej priemernej MES je znázornená ako funkcia priemernej hustoty CPG, hustota génu (počet génov na Mb), LINE množstvo (dĺžka LINE na Mb) a SINE množstvo (dĺžka SINE za mb) pre každého chromozómu. (B) V prípade chromozómu 22, priemerná hustota CPG (tieňované sivé) a MES (čierna krivka) boli získané v 1 Mb posuvné okná. Polohy prepísaných génov (čierne pruhy v spodnej časti), CG ostrovy (modré pruhy hore) a dlhé zaznie (viac ako 1 kb, červené pruhy hore) sú porovnané na pozadí metylácie DNA a hustote CPG ( vľavo). Priemerné MES pre CGI, génovej subjekty a opakovanie (vpravo). (C) Rozloženie génových orgánov a CGI MES (vľavo). Priemerné MES pre promótorom asociované a promótorom nezávislé CGI je znázornené na pravej strane. (D) Priemerné MES pre promótorom podskupín, založené na existencii CGI (vľavo). (E) Základné informácie o intergenové, exonic a IntronA oblastiach, v závislosti na dĺžke, počet CPG, a mapované číta (vľavo). Distribúcia intergenové, exonic a intronové MES je znázornené na pravej strane. (F) Základné informácie o vstupnom 1-kb oblasti, 5 'UTR exóny, kódujúce exóny, 3' UTR exóny a downstream 1-kb región podľa dĺžky, počtu CPG a mapované číta (vľavo). Distribúcia MES pre každý prvok je znázornené na pravej strane.
Všeobecne platí, že CGI majú tendenciu zostať bez metylácie v normálnej tkanive. Analyzovať vysoká metylácie vzory CGI, sme skontrolovali priemernú distribúciu MES a našiel mierne bimodálnej obrazec (obrázok 1C). Asi 66% (11.376 /17.284) z CGI v ľavom vrchole prekrývali s promótorom (1 kb od našej definície). Naproti tomu 13% (1386/10357), z CGI v pravom vrchole prekrývali s promótorom, čo naznačuje, že väčšina promótor spojený CGI sú nemethylované. Na rozdiel od promotora súvisiace s CGI, promótor nezávislé CGI bol ťažko methylata (obrázok 1C). Hoci väčšina promótory CGI-pozitívne neboli methylata, CGI-negatívne promotory vykazovali relatívne vysoké úrovne metylácie (obrázok 1D). Zároveň kontroluje úroveň metylácie promotorov hustotou CPG, ako bolo definované vyššie [38] (Ďalší súbor 3: Tabuľka S4). Metylácia vzor promotérov bola nepriamo úmerná hustote CPG (ďalší súbor 2: Obrázok S4). Na druhej strane, génovej tela CGI obsahujúce mali vyššie hladiny, ako sú metylačnej bez CGI (obrázok 1D).
Ďalšie sme analyzovali metylačnej obohacovanie vzory v rôznych komentovaný genomových elementov oblasť oblasti, ktoré boli prednostne methylata. Génový oblasti zaberajú asi 40% ľudského genómu, ale asi 53% z celkového počtu prečíta je v tejto oblasti, s väčšinou číta, že sa nachádza v oblasti intronom (Tabuľka 1). Aj keď významná časť metylovaných fragmentov spadá do IntronA oblastiach, je pomer mapovaná číta na dĺžku exónov je podstatne vyššia ako u IntronA, čo naznačuje, že exóny sú vysoko methylata ako IntronA (obrázok 1E). V rámci regiónov génov asociovaných s, obohatenie kódujúcich exónov je dokonca vyššia ako u iných regiónoch Ako bolo oznámené predtým (obrázok 1F a tabuľka 2) [39]. To silne naznačuje, že metylácie hrá úlohu v exóne 1 regulation.Table ľudskom genóme a normálne informácie vzorky génovej a intergenové oblasti
Human Genome informácie
Normal Sample informácie
relatívny pomer obohatenia
funkčné kategórie
Dĺžka (bp)
pomer
# CPG
Ratio
Číta
Ratio
vs. Dĺžka
vs. CPG Count
Genic
1,184,139,094
39.46
13,262,253
47.09
20,854,434
53.25
1.35
1.13
Exon
68,035,894
2.27
1,808,089
6.42
4,350,405
11.11
4.90
1.73
Intron
1,122,817,725
37.41
11,613,113
41.23
17,358,273
44.32
1.18
1.07
Intergenic
1,816,976,186
60.54
14,901,610
52.91
18,310,273
46.75
0.77
0.88
Human Genóm
3001115280
100
28163863
100
39164707
100
1,00
1,00
Tabuľka 2 Ľudský genóm a normálne informačné vzorka génu anotovaných regiónov
Human Genome informácie
Normal Sample informácie
relatívny pomer obohatenia
Funkčné Kategória
Dĺžka (bp)
pomer
# CPG
Ratio
Číta
Ratio
vs. Dĺžka
vs. CPG grófa
spoločností upstream 1 KB
24468069
0,82
937.748
3,33
535.593
1,37
1,68
0,41
5'UTR exónov
8436529
0,28
411.563
1,46
292.654
0,75
2,66
0,51
kódujúcich exóny
33384619
1,11
1077913
3,83
3448755
8,81
7,92
2,30
3'UTR exónov
28387978
0,95
378.012
1,34
806.832
2,06
2,18
1,53
Následný 1 kb
23136263
0,77
340.866
1,21
551.071
1,41
1,83
1,16
Human genóm
3001115280
100
28163863
100
39164707
100
1,00
1,00
Zmeny metylácie DNA vzory spojené s rakovinou žalúdka
Keď priemerná chromozomálne MES z methylome rakoviny bolo v porovnaní s kontrolným tkaniva, sme zistili, že všetky chromozómy v tkanive rakoviny inklinoval byť hypomethylated (dodatočný súbor 2: Obrázok S5). S výhľadom chromozomálnych celé bolo zistené, že CGI bohaté regióny, ktoré majú byť špecificky hypermethylated, zatiaľ čo opakované bohaté oblasti boli široko hypomethylated (obrázok 2A; Ďalší súbor 2: Obrázok S6). Analyzovať zmeny metylácie genómovej elementy, vyrovnané sme každý prvok v počiatočných a koncových miest a potom získala priemerné MES v každom príslušnom mieste. Pozoruhodné je, že sme zistili, hypermetylace v hornej oblasti, a to najmä od 500 bp proti smeru k počiatku transkripcie (obrázok 2B). To je v súlade s hypermetylace z promotorových oblastí často pozorované u rakoviny. Obrázok 2 Porovnanie metylácie vzorov v normálnej a rakovinové tkanivá. (A) Priemerné MES krivka pre normálne (čierna) a rakovinové (červená), tkanivá v chromozóme 19 (vľavo). Priemerné MES pre CGI, génovej subjekty a opakovanie (vpravo). (B) metylácie DNA génových komentovaný prvkov. Každý prvok (proti prúdu 1 KB, exón, intronom, a za 1 kb) bol rozdelený do 20 zásobníkov a priemerné MES sa získa pre každý bin všetkých zodpovedajúcich prvkov. (C) metylácie DNA na koncoch transkriptov a kódovanie koncov región. Priemerné MES bolo získané vo posuvné 50-bp okna podľa jeho vzdialenosť od začiatku prepisu (prvý) a koniec (druhý) pre CGI-pozitívnej promótory, ako aj na začiatku transkriptov (tretia) a na konci (štvrtom) pre CGI- pozitívne predkladatelia. (D) DNA metylácie celkom 5 'UTR exónov (vľavo) a 5' UTR kódujúcich exónov (vpravo).
Oblasť so stredom v počiatku transkripcie ukázalo úplne rôzne vzory v závislosti na prítomnosti CGI, čo odráža nízku stav metylácie promotorov CGI obsahujúce (Obrázok 2C). Tiež sme zistili, že v rakovinové tkanive, pozoruhodné hypermetylace promotorov CGI obsahujúce objavia, a že hustota CPG je rozhodujúca pre zvýšenie metylácie DNA (obrázok 2C). Pre ďalšiu analýzu, či 5 'oblasti génov boli hypermethylated podobne ako promotérov génov, Skontrolovali sme metylácie vzoru prvých exónov. Zaujímavé je, že sme zistili, že prvý exón bol hypermethylated iba vtedy, ak bol 5'-koniec kódujúci exon, ale nie, keď to bolo 5 'UTR exon (obrázok 2D). Tieto regióny tiež obsahoval vysokej hustote CPG. Preto sa CGI na upstream oblastí génov, promótor, a začať písať kód sa zdajú byť hlavnými cieľmi DNA hypermetylace pri liečbe rakoviny.
Metylácia vzor CPG ostrovov
Ak chcete preskúmať vzájomný vzťah medzi umiestnením CGI a metylácie DNA, my subgrouped CGI v závislosti na ich pozíciu v rámci genómu. Konkrétne, boli rozdelené do kategórií ako 5 '(ktorá sa nachádza medzi 1 kb proti prúdu a počiatočného miesta kódovania génu), intragenic (intragenic CGI mimo 5' konci), a intergenové (nachádzajúce sa v non-génovú oblasť) (ďalšie súbor 1: tabuľka S5). Hoci hustota CPG bol podobný medzi týmito troma skupinami, non-5 'CGI (intragenic a intergenový CGI) boli významne viac ako methylata 5' CGI (ďalší súbor 2: Obrázok S7). Ďalej sme porovnávali priemerné MES subgrouped CGI a zistil, že metylácie všetky CGI bol všeobecne zvýšená. Avšak, relatívna rozdiel MES naznačili, že zmena v metylácie v 5 'CGI bola významne vyššia než u ostatných CGI (obrázok 3A), čo odráža dôležité role 5' CGI v rakoviny. Rozsah 5 'CGI hypermetylace významne koreluje s presahom počiatku transkripcie (obrázok 3B). Obrázok 3 DNA metylácia CPG ostrovov. (A) Relatívna diferenciálnej MES o subgrouped CGI. (B) Korelácia medzi diferenciálnej CGI metylácii a vzdialenosť k počiatku transkripcie. (C) Korelácia medzi úrovňou génovej expresie a hypermetylace z CGI. (D) Metylácia špecifické PCR histonových génov, ktorý má najvyššiu diferenciálnej MES hodnoty. . M1 a U1 zodpovedajú HIST3H2A, zatiaľ čo M2 a U2 zodpovedajú HIST3H2B
Ak chcete preskúmať funkcie génov podstupujúcich diferenciálnej metylácie na 5 'CGI, my vybraných génov s vysoko diferenciálnej CGI MES (MES diferenciálnej > 1). Potom sme vykonali génovej ontológie (GO) Analýza získať vhľad do mechanizmov zodpovedných pri karcinóme (tabuľka 3). Keď boli gény zoskupené do rôznych kategórií GO, sme zistili, že HOx
génové klastre a nucleosome génové klastre montážne súvisiace boli ciele pre hypermetylace, zatiaľ čo apoptózy súvisiace s génových klastrov boli ciele pre hypometylácii. Je zaujímavé, že naše zistenia, že HOx
génových klastrov boli preferenčné ciele pre metylácie DNA je v súlade s predchádzajúcou správou [40]. Okrem toho génovej pozemky potvrdil, že hypermetylace bol CGI-špecifické rakoviny (ďalší súbor 2: Obrázok S8). Odhadnúť zmeny v expresných vzoroch spôsobené hypermetylace 5 'CGI sme vykonali funkčné analýzu génovej expresie údajov získaných z cDNA microarray experimentov. Hypermetylace 5 'CGI bol významne koreluje s down-reguláciu génov (p
= 0,03) (obrázok 3C ďalšie súbor 3: Tabuľka S6 a S7). To znamená, že umlčanie génov metylácie môže byť priamo ovplyvnená stupňom hustoty CPG a 5 'CGI hypermetylace. Analyzovali sme DNA metylačného statusu génov s hypermethylated 5 'CGI a downregulated expresné vzory. Medzi tieto bol gén kódujúci typ Histon H2B 3-B (HIST3H2BB
). Analýza HIST3H2BB
metylácie promótorom pomocou metylácie-špecifické PCR bolo zistené, že väčšina pacientov s rakovinou (8/10, 80%), vykazovali zvýšenú metylácie v oblasti promótorom (obrázok 3D) .Table 3 Funkčné anotácie zhlukovaniu génov s hypermethylated 5'CGIs
Cluster Anotácia 1
Obohacovanie skóre: 3,27
Count
P_Value
GOTERM_BP_FAT
nucleosome montáž
11
3.90-04
GOTERM_BP_FAT
chromatínu montáž
11
5.20-04
GOTERM_BP_FAT
proteín-DNA komplexu montážne
11
7.40-04
Anotácia Cluster 2
Obohacovanie skóre: 2,92
Count
P_Value
Interpreti
histónov jadra
8
6.80-04
SP_PIR_KEYWORDS
nucleosome jadro
8
8.60-04
GOTERM_CC_FAT
nucleosome
8
3.10-03
Anotácia Cluster 3
Obohatenie skóre: site
17
5.10E-03
INTERPRO
Homeobox
17
5.70E-03
SP_PIR_KEYWORDS
Homeobox
17
5.80E-03
INTERPRO
Homeodomain-related
17
6.40E-03
SMART
HOX
17
1.40E-02
D4
3
9.10E-03
SP_PIR_KEYWORDS
embryo
3
3.30E-02
PIR_SUPERFAMILY
PIRSF002612:homeotic
 Štúdia objasňuje príčiny oslabujúcej bolesti čriev
Štúdia objasňuje príčiny oslabujúcej bolesti čriev
 Dezinfekčné prostriedky pre domácnosť by mohli prispieť k riziku obezity u detí
Dezinfekčné prostriedky pre domácnosť by mohli prispieť k riziku obezity u detí
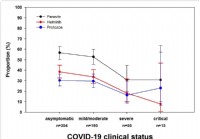 Výskum ukazuje, že zamorenie črevnými parazitmi znižuje závažnosť ochorenia COVID-19
Výskum ukazuje, že zamorenie črevnými parazitmi znižuje závažnosť ochorenia COVID-19
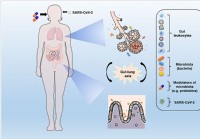 Mikrobiotická modulácia a obnovenie eubiózy by mohli pomôcť obmedziť komplikácie súvisiace s COVID-19
Mikrobiotická modulácia a obnovenie eubiózy by mohli pomôcť obmedziť komplikácie súvisiace s COVID-19
 Krajiny so staršou populáciou majú vyššie infekcie a úmrtia na SARS-CoV-2,
Krajiny so staršou populáciou majú vyššie infekcie a úmrtia na SARS-CoV-2,
 Vedci identifikovali baktériu s aktivitou anti-SARS-CoV-2 in vitro:Dolosigranulum pigrum
Vedci identifikovali baktériu s aktivitou anti-SARS-CoV-2 in vitro:Dolosigranulum pigrum
 Veľká štúdia uvádza, že vírusová záťaž SARS-CoV-2 je najnižšia u detí
Táto provokatívna nová štúdia výskumníkov v Holandsku potvrdzuje hypotézu, že v prípade závažného akútneho respiračného syndrómu koronavírusu 2 (SARS-CoV-2) infekcia vyšší vek je spojený so zvýšenou v
Veľká štúdia uvádza, že vírusová záťaž SARS-CoV-2 je najnižšia u detí
Táto provokatívna nová štúdia výskumníkov v Holandsku potvrdzuje hypotézu, že v prípade závažného akútneho respiračného syndrómu koronavírusu 2 (SARS-CoV-2) infekcia vyšší vek je spojený so zvýšenou v
 Nový superaktivujúci makrofágový receptor by mohol vysvetliť hyperzápal pri závažnom ochorení COVID-19
Imunita je zaujímavá vec. Aj keď sú nevyhnutné pri ochrane tela pred napadnutím patogénmi a cudzími antigénmi, môže sa tiež obrátiť proti telu a spustiť deštruktívne imunologické procesy. Nová štúdia
Nový superaktivujúci makrofágový receptor by mohol vysvetliť hyperzápal pri závažnom ochorení COVID-19
Imunita je zaujímavá vec. Aj keď sú nevyhnutné pri ochrane tela pred napadnutím patogénmi a cudzími antigénmi, môže sa tiež obrátiť proti telu a spustiť deštruktívne imunologické procesy. Nová štúdia
 Geneticky ladiace črevné baktérie znižujú riziko rakoviny hrubého čreva a konečníka u myší
Vedci zistili, že úprava génov baktérií prítomných v črevách myší by mohla pomôcť znížiť zápal a súvisiace riziko rakoviny hrubého čreva a konečníka. Výskum vedcov z UT Southwestern bol zverejnený ten
Geneticky ladiace črevné baktérie znižujú riziko rakoviny hrubého čreva a konečníka u myší
Vedci zistili, že úprava génov baktérií prítomných v črevách myší by mohla pomôcť znížiť zápal a súvisiace riziko rakoviny hrubého čreva a konečníka. Výskum vedcov z UT Southwestern bol zverejnený ten