Identifisering av DNA metylering endringer knyttet til menneskelig magekreft
Abstract
Bakgrunn
epigenetisk endring av genuttrykk er en vanlig hendelse i kreft hos mennesker. DNA metylering er en velkjent epigenetisk prosess, men bekrefter det nøyaktige innholdet i epigenetiske endringer knyttet til kreft er fortsatt vanskelig.
Metoder
Vi profilert den methylome av menneskelig magekreft vev ved 50-bp oppløsning ved hjelp av en metylert DNA berikelse teknikk (metylert CpG island utvinning analyse) i kombinasjon med et genom analysator og en ny normalisering algoritmen.
Resultater
Vi var i stand til å få en helhetlig oversikt over arrangører med ulike CpG tettheter, inkludert CpG Øyene (CGIer), transkripsjon organer, og ulike gjenta klasser. Vi har funnet at magekreft var forbundet med hypermethylation av 5 'CGIer og 5'-enden av kodings exoner samt hypometylering av gjentatte elementer, slik som korte utblandet kjerneelementene og det sammensatte element SVA. Hypermethylation av 5 'CGIer ble signifikant korrelert med nedregulering av tilhørende gener, slik som de i HOX
og histon genfamilier. Vi har også oppdaget langtrekkende epigenetisk Silencing (LRES) regioner i magekreft vev og identifisert flere hypermethylated gener (MDM2
, DYRK2
, og lyz
) innen disse områdene. Metylering status for CGIer og genet merknader elementer i metastatiske lymfeknuter var en mellomting mellom normalt og kreftvev, noe som indikerer at metylering av spesifikke gener økes gradvis i kreftvev.
Konklusjoner
Våre funn vil gi verdifulle data for fremtidig analyse av CpG metylering mønstre, nyttige markører for diagnose av magekreft, samt en ny analysemetode for kliniske Epigenomics undersøkelser.
Bakgrunn
mage~~POS=TRUNC kreft~~POS=HEADCOMP er den nest største årsaken til kreft dødsfall på verdensbasis etter lungekreft, noe som resulterer i mer enn 800.000 dødsfall på verdensbasis hvert år [1]. Dagens 5-års overlevelse av personer diagnostisert med magekreft er bare 20-30%, med lav rente som kan tilskrives det faktum at de fleste tilfeller allerede er i et avansert stadium når diagnosen. Som i alle kreftformer, forblir tidlig påvisning den mest lovende metode for forbedring av overlevelsesraten. Derfor forstå årsaken til tumorigenesis i menneskets mage vev er viktig.
Infeksjon med H. pylori
er et veletablert og vanlig årsak til magekreft. Imidlertid, endringer i forskjellige genetiske faktorer er også viktig for å øke magekreftrisiko. Det er vel kjent at kromosomal ustabilitet som stammer fra genetiske faktorer så som mikrosatelitt ustabilitet, så vel som KRAS
og p53
mutasjoner resultere i utvikling av svulster. Flere genomiske studier har identifisert germline mutasjoner i spesifikke gener [2-4] og sykdom utsatt loci [5, 6] for magekreft. Nyere studier som sammenligner magekreft og normalt vev har identifisert en rekke genetiske markører, inkludert diagnostiske markører [NF2 product: [7], INHBA product: [8], SFRP4 product: [9]], prognostiske markører [CD9
[10], CDH17 product: [11], PDCD6 product: [12]], og magekreft knyttet gener [MUC13 product: [9], CLDN1 product: [13], Ki67
og CD34 product: [14]]. I tillegg har epigenetiske mekanismer som DNA-metylering og histonmodifikasjonene blitt funnet å være viktige i reguleringen av ekspresjonen av gener som er involvert i biologi og sykdom i mage-tarmkanalen [15].
DNA-metylering spiller en avgjørende rolle i eukaryoter og er forbundet med en rekke viktige mekanismer, inkludert genomisk preging, X-kromosom inaktivering, aldring, og karsinogenese. Endring av DNA metylering i genomet er funnet i nesten alle typer kreft, og kan føre til endringer i genuttrykk, for eksempel over-uttrykk for onkogener og stanse av tumorsuppressorgener under kreftutvikling [16]. Flere studier har vist at opphopning av genetiske og epigenetiske forandringer i mage forstadier til kreft kan påvirke et stort antall mål, som for eksempel DNA-reparasjonssystemkomponenter, tumor suppressors, onkogener, cellesyklus regulatorer, vekstfaktorer og adhesjonsmolekyler [17-20] . Imidlertid har disse studier er primært fokusert på et par kandidatgener eller dekket bare en del av hele genomet. Dermed tilgang til en global oversikt over de epigenetiske endringer knyttet til kreftutvikling har vært vanskelig. Spesielt forståelse DNA metylering endringer i intragenic regioner, CpG øyer, intergeniske regioner, og gjenta sekvenser fortsatt begrenset. Derfor er det stor interesse for genom-wide analyse av avvikende DNA metylering i disse regionene.
For omfattende genom-skala profilering av DNA metylering i embryogenese og karsinogenese, høyoppløselige hele genomsekvensering metoder som BS-seq [21 -24], har MeDIP-seq [25, 26], og MethylCap-seq [27-29] blitt utviklet. Til tross for den raske utviklingen av sekvensebasert kartteknologi, er det fortsatt mangel på komparativ forskning, som er avgjørende for kliniske Epigenomics studier, inkludert de fokusert på kreft. I motsetning til microarray-baserte tilnærminger, blir sekvense data produsert i et format som ikke er mottagelig for differensiell analyse, og analysen arbeidsflyten har ikke blitt standardisert. Derfor er beregningsmessig rimenormaliserings metoder for å håndtere beregnings byrden av å behandle store størrelse, høy oppløsning sekvensering av data.
Her introduserte vi en normalisering algoritmen, som tar hensyn til den prøven spesifikke total lese tetthet, den romlige fordelingen av CpG loci, og bakgrunn sekvense skjevhet. Deretter har vi laget en omfattende hel-genom methylome av normal mage vev, magekreft vev, og metastatiske lymfeknuter bruker MethylCap-seq metode og innhentet detaljert informasjon om sin uro under kreftutvikling og metastasering. Dette er lett anvendelig til en komparativ analyse av methylomes og andre typer epigenomic data, og det har særlig betydning for kliniske Epigenomics.
Metoder
Gastric vevsprøver
Vi oppnådd tre snap-frosne mage svulster og matchet normal gastrisk vev fra Seoul National University College of Medicine for methylome studien. I tillegg ble tjueåtte matchet par av normale og kreftmage vev oppnådd for ytterligere bekreftelse. Alle prøver ble oppnådd ved endoskopisk reseksjon under behandlingen av pasientene som ga informert samtykke
metylert DNA gjenvinning assay (MIRA)
Genomisk DNA fra 25 mg av gastrisk vev ble renset ved hjelp av DNeasy Blood &.; Tissue Kit (Qiagen, Valencia, CA). Genomiske DNA-prøver fra 3 individer ble samlet ved samme konsentrasjon. MIRA ble utført som beskrevet tidligere [30-32]. Kort, GST-merket MBD2b og Hans-merket MBD3L1 proteiner ble fremstilt som beskrevet. 15 pg av genomisk DNA ble fragmentert til 100 ~ 500 bp ved ultralydbehandling og inkubert med 28 ug av renset GST-MBD2b protein, 28 ug av His-MBD3L1 protein og 7 ug av JM110 Bakteriell DNA i 6 timer. 30 ul av MagneGST kuler (Promega, Madison, WI) preblocked med 7 ug av JM110 bakterielle RNA ble tilsatt og inkubert ved 4 ° C med rotering i 45 minutter i slutt 600 ul av MIRA-bindingsreaksjonsblandingen. Perlene ble vasket tre ganger med 1 ml vaskebuffer, og metylerte fragmenter ble eluert ved inkubering ved RT i 5 minutter og deretter 56 ° C i 30 minutter med 30 pl av TE inneholdende RNase A (100 ug, Qiagen) og Proteinase K ( 15 ug, Qiagen). Eluerte DNA-fragmenter ble ytterligere renset ved hjelp Qiaquick PCR rensing kit (Qiagen).
Illumina Genome Analyzer sekvense
Vi brukte 10 ng av eluert DNA for Illumina Genome Analyzer sekvensering. Etter ligering av et par Solexa adaptere, Ligeringsprodukter med den maksimale innskuddsstørrelse på 200 bp ble gelrenset på 2% agarose, og utsatt for PCR-amplifikasjon. Cluster generasjon og 36 sykluser av sekvensering ble utført i henhold til produsentens instruksjoner. Vi sekvensert 120 ul av adapter-ligert, størrelse-fraksjonert DNA (2 ~ 16:00) på Illumina Genome Analyzer. Sekvens koder ble kartlagt for det menneskelige genom (UCSC hg18 database basert på NCBI Bygg 36,1 montasje) bruker Solexa Analyse Pipeline (versjon 0.3.0). Sekvensert lesninger av 34 bp (med unntak av første og siste nukleotid) som passerte kvalitetskontroll Filtrene ble anvendt.
Databehandlings og MES beregning
Vi utvidet 3'-enden av det 34 bp-leser ved 200 bp for å dekke DNA fragmenter som er bundet av MBD proteiner. Den avlesning ble konvertert til nettleseren utvidbar data (BED) filer for visualisering i UCSC genom nettleser http:. //Genom UCSC edu /.. Vi telte overlappende sekvens koder på 50 bp oppløsning. For å finne anrikede genomiske regioner, ble antallet kartlagt står i et glidende vindu med en kb sammenlignet med det totale antall leser eller bakgrunnen antall står i genomet. Som sådan, ble MES beregnes på to måter; en er som log2 av (target lese telle /target størrelse) /(total lese- teller /genomstørrelse) og angis til null, er den andre som den log2 av (target lese telle /target størrelse) /(bakgrunn lese telle /bakgrunn størrelse) og lysekroner til null. For å justere for bakgrunnen sekvense bias, ble MESbg beregnes på samme måte for innspill sekvense uten affinitet rensing og trekkes fra MES.
Genomisk stillinger CGIer, arrangører, avskrift organer, CDS, og repeterende elementer
Alle genomiske stillinger CGIer, utskrifter og gjenta elementene ble lastet ned fra UCSC genom nettleser. Totalt 27 639 CGIer (bortsett fra tilfeldig plassert CGIer) ble forutsagt ved hjelp av følgende kriterier: GC-innhold på 50% eller større, lengde større enn 200 bp, og forhold som er større enn 0,6 fra observerte antall CpG-dinukleotider til det forventede antall [33] . NCBI mRNA referansesekvenser samling (RefSeq fra versjonen 46; 11 mars 2011) ble brukt for å identifisere transkripsjonsenheter med den definerte transkripsjon start, end-områder og CDS starte, endesider. For arrangører, brukte vi den regionen 500 bp oppstrøms ~ 500 bp nedstrøms for transkripsjon start stedet. Vi fikk ~ 5 millioner gjenta steder som hadde blitt bestemt av RepeatMasker program basert på RepBase bibliotek av gjentakelser.
Metylering nivå av genomiske elementer
metylering nivå av et CGI, promoter, gen-kropp, og gjenta element ble estimert ved hjelp av overlappende MES hvert element. MES = 0 ble brukt til å definere unmethylated elementer. For å måle hypermethylation eller hypometylering i kreft, vi beregnet differensial rotet som (Cancer MES - Normal MES). Differential MES > 1,0 ble anvendt som en terskel. For å forstå funksjonene til utvalgte gener, brukte vi ontologi klassifisering av gener gjennom DAVID Functional merknad Clustering verktøy http:.... //David abcc ncifcrf gov /
genekspresjonsanalyser
microarray produkt som brukes i denne studien var Code Menneskelig Whole Genome 55 K chip (GE Healthcare, USA). Alle eksperimentelle prosedyrer inkludert cRNA mål forberedelse, hybridisering, ble post-hybridisering dye kopling utføres ved hjelp av leverandørens anbefalte protokoller. Resultat filene ble importert til GeneSpring GX 7.3 (Agilent Technologies, USA) for filtrering og grunnleggende statistisk analyse. Blant 55 K gener på microarray, vil bare de gener med foreliggende flagg i minst 50% av prøvene ble utvalgt for etterfølgende analyse. Microarray data ble avsatt på GEO http:.. //Www NCBI NLM nih gov /geo /(tiltredelse antall GSE33651)
MIRA og real-time qPCR
MIRA... ble foretatt på ytterligere fire individuelle prøver. DNA ble renset fra supernatanten og overvåkes av sanntids qPCR ved bruk av Roche 480 maskin. Sekvensene av brukte primere er presentert i tilleggsfiler. 1: Tabell S1
Bisulfite behandling, metylering spesifikk PCR og pyrosekvensering
Vi isolert genomisk DNA fra individuelle prøven ved hjelp av en Qiagen DNeasy Tissue Kit (Qiagen). Bisulfite behandling ble utført ved hjelp av EZ DNA metylering gull kit (Zymo forskning) i henhold til produsentens instruksjoner. Bisulfitt-behandlede DNA ble lagret ved -80 ° C inntil videre anvendelse. Primere som brukes for MSP ble utformet ved hjelp Methprimer [34], og er vist i tilleggsfiler 1: Tabell S1. PCR ble utført med HotStarTaq polymerase (Qiagen) og inkludert en første inkubasjon ved 95 ° C i 15 min, etterfulgt av 40 sykluser på 95 ° C i 1 min, 59 ° C i 1 minutt og 72 ° C i 40 sekunder, etterfulgt av en syklus på 72 ° C i 10 minutter. MSP-produkter ble separert på 2% agarosegeler og visualisert ved EtBr-farging. De pyrosekvensering reaksjoner ble automatisk utført med en PSQ 96 system (pyrosekvensering AB) i henhold til produsentens instruksjoner. I korte trekk ble biotinylert PCR-produkt (50 ul) ble renset ved hjelp av streptavidin-sepharose-kuler (Amersham Biosciences). Det rensede produkt ble lastet inn i reagensen patronen med enzymet, substratet og dNTP inkludert i PSQ96 SNP Reagent Kit (pyrosekvensering AB). De sekvense primere for pyrosekvensering er vist i tilleggsfiler. 1: Tabell S1
Resultater
Behandling av MIRA-seq methylome data
Vi renset metylert DNA beriket gjennom MIRA (metylert CpG island utvinning analyse) og sekvensert DNA ved hjelp av neste generasjons sekvensering. DNA-metylering-nivåer ble bestemt ved hjelp av sekvensering lese tellinger av de tilsvarende regionene, 50 BP intervaller, som beskrevet i avsnittet Metoder. Vi skapte DNA metylering kart for både normale og kreft mage vev. For hver prøve, fikk vi ca 10 millioner sekvens leser (tilleggsfiler 1: Tabell S2). Hver methylome inneholdt ~ 140 millioner CpG leser, som dekker ~ 48% av alle genomiske CpG områder unntatt sentromerer (Tilleggs fil 1: Tabell S3). Den gjennomsnittlige dekningen av CpG leser i hvert methylome var 4,5X. Til støtte for den høye følsomheten MIRA, genomiske segmenter som bare inneholder en CpG hadde høyere lese teller enn de uten CpG (p
verdi = 0), noe som tyder på at enkelt CpG endringer kan løses ved hjelp av MIRA. Den gjennomsnittlige sekvens leser økte proporsjonalt med antall CPGs innenfor et 50-bp intervall, og i virkeligheten, MIRA dekningen ikke var lav, selv i områder med lav tetthet CpG (Tilleggs fil 2: Figur S1). Samlet utgjør disse resultatene viser at MIRA var vellykket i å utvinne en tilstrekkelig andel av denaturert regioner. Som for nøyaktigheten av MIRA, ~ 99% av MIRA-fanget fragmenter hadde minst en CpG området innenfor deres rekkefølge, noe som indikerer en lav falsk treffprosent.
Å måle anrikning av lokale metylering signaler beregnet vi metylering berikelse score (rot ) ved å skaffe et skrive telling i en gitt region, og deretter utføre normalisering for å kontrollere for det totale lese telle (MEST) i prøven (global normalisering) eller den lokale lese count (MESl) i en brukerdefinert omliggende regionen (lokal normalisering) (se Methods). Dette muliggjør en direkte sammenligning av uavhengige prøver med forskjellig tetthet lest. Vi utførte deretter en logaritmisk transformasjon av den deriverte stillingen. Sammen med å ha andre matematiske fordeler, gir dette den fordelen av variansen stabilisering, spesielt for høy lese tellinger, som ofte er kombinert med høye tekniske variasjoner som kan innføre betydelig skjevhet i dataene.
Vi vurderte den statistiske signifikans av MES i to veier. Randomisert rotet ble generert numerisk ved en permutasjon av genomiske stillinger fra vår sekvens leser. Bakgrunns MES (MESbg) ble eksperimentelt oppnådd ved sekvensering av den normale genomet uten affinitetsrensing. Som forventet, de reelle data ga markant høyere berikelse score (tilleggsfiler 2: Figur S2). Spesielt MESbg var høyere enn MES fra randomiserte genomer, en indikasjon på at bakgrunns sekvenser alene kan skape berikelse, sannsynligvis på grunn av kromatin tilgjengelighet og forsterkning bias. I samsvar med de siste rapportene [35], illustrerer dette behovet for en skikkelig kalibrering for iboende sekvense bias. Derfor har vi normalisert våre MES med MESbg.
For å finne den optimale tilstand for normalisering, sammenlignet vi den statistiske egnethet av ulike normaliseringsmetoder. Tag fordeling langs genomet kan modelleres ved Poissonfordelingen [36, 37]. Den egnethets ble testet ved hjelp av Kolmogorov-Smirnov test. I denne testen indikerer en lav D statistikk en god passform. Mens Poisson modellen gjorde det bedre enn Gaussian samlet, MES viste en bedre passform enn rå lese tellinger (tilleggsfiler 2: Figur S3), illustrerer sjelden hendelse natur log-skalert lese teller mål. De normaliserte MESl kalibrert av kontroll sekvensering (MESbg) ga enda bedre resultater enn normalisert MEST kalibrert av kontroll sekvensering (MESbg).
Global og kromosom utsikt over DNA metylering
etter å ha bekreftet den beste metoden for poeng genom-wide metylering nivåer 50-bp mellomrom, må vi først undersøkte kromosom metylering mønstre av normale prøver. De gjennomsnittlige MES beregnet for hvert kromosom antydet at CpG-rikt og gen-rik kromosomer har en tendens til å være sterkt metylert (figur 1A). De metylering nivåer av kromosomer med store mengder lange ispedd kjerneelementer (linjer) var relativt lav (f.eks kromosom 4). Interessant er mengden av korte utblandet kjerneelementer (sinus) var proporsjonal med kromosomet metylering mønster. Dette er sannsynligvis forårsaket av det faktum at sinus blir vanligvis gruppert i gen-rike regioner. Kjønnskromosomer ble globalt hypomethylated med lavere CpG tetthet og høyere gjenta innhold enn autosomer. Siden vi brukte vev tatt fra en hann i dette eksperiment, er den globale hypometylering av X-kromosomet observeres ikke er forbundet med X-inaktivering. Kromosom bred utsikt rekapitulert høy CpG tetthet og høy metylering rundt gen-rik (se svarte striper nederst) og CGI-rike regioner (se blå striper øverst) (figur 1B). I kontrast, ble lav CpG tetthet og lav metylering observert rundt gen fattige regioner som var rik på langtrekkende gjentar (> 1 kb) (se røde striper øverst). De gjennomsnittlige MES tyder på at nivået av metylering CGIer som er betydelig høyere enn den til fremkallende regioner eller gjentagelser (figur 1B). Figur 1 metylering mønstre av normal mage vev. (A) Kromosom-wide gjennomsnittlig MES er vist som en funksjon av gjennomsnittlig CpG tetthet, genet tetthet (antall gener per Mb), LINE mengde (lengden av LINE per Mb), og SINE mengde (lengden av SINE per mb) for hvert kromosom. (B) For kromosom 22, ble den gjennomsnittlige CpG tetthet (gråfarget) og MES (svart kurve) oppnådd i en MB skyvevinduer. Posisjonene til transkriberte gener (svarte striper nederst), CG øyer (blå striper øverst), og lange repetisjoner (> 1 kb, røde striper øverst) sammenlignes på bakgrunn av DNA metylering og CpG tetthet ( Igjen). De gjennomsnittlige MES for CGIer, Gene organer, og gjentar (til høyre). (C) Fordelingen av genet organer og CGI MES (til venstre). De gjennomsnittlige MES for promoter-forbundet og promoter uavhengig CGIer er vist til høyre. (D) De gjennomsnittlige MES for promoter undergrupper, basert på eksistensen av CGI (til venstre). (E) Grunnleggende informasjon om intergeniske, exonic, og intronic regioner, ifølge lengde, CpG nummer, og kartlagt leser (til venstre). Fordelingen av intergeniske, exonic, og intronic rot er vist til høyre. (F) Grunnleggende informasjon på oppstrøms 1-kb region, 5 'UTR eksoner, koding eksoner, 3' UTR eksoner, og nedstrøms en-kb region i henhold til lengde, CpG nummer og kartlagt leser (til venstre). Fordelingen av MES for hvert element er vist til høyre.
Vanligvis CGIer tendens til å forbli metylering gratis i normalt vev. For å analysere høy metylering mønstre av CGIer, sjekket vi den gjennomsnittlige MES distribusjon og fant en litt bimodal mønster (figur 1C). Om 66% (11 376/17 284) av CGIer i venstre topp overlappet med en promoter (1 kb etter vår definisjon). I motsetning til 13% (1386/10 357) av CGIer i riktig peak overlappet med en promoter, noe som tyder på at de fleste promoter-forbundet CGIer er unmethylated. I motsetning til arrangøren relaterte CGIer, arrangøren uavhengig CGIer ble tungt metylert (figur 1C). Selv om de fleste CGI-positive arrangører ikke ble metylert, CGI-negative arrangører viste relativt høye metylering nivåer (figur 1D). Vi har også sjekket metylering nivået av arrangører av CpG tetthet som tidligere definert [38] (tilleggsfiler 3: Tabell S4). Metylering mønster av arrangører ble omvendt relatert til CpG tetthet (tilleggsfiler 2: Figur S4). På den annen side, CGI-inneholdende genet legemer hadde høyere metylering nivå enn de uten CGIer (figur 1D).
Deretter vi analyserte metylering berikelse mønstre ved forskjellige kommenterte genomiske elementer for å utforske regioner som ble fortrinnsvis metylert. Skapte regioner opptar ca 40% av menneskelige genom, men om lag 53% av den totale leser er innenfor denne regionen, med de fleste leser blir plassert i intronic regionen (tabell 1). Selv om en betydelig andel av denaturert fragmenter faller innenfor intronic regioner, forholdet mellom tilordnet leser til lengden av exoner er betydelig høyere enn det for introner, som tyder på at exoner er mer sterkt metylert enn introner (figur 1E). Innenfor gen-assosiert regioner, berikelse av kodende eksoner er enda høyere enn for andre regioner som tidligere er rapportert (figur 1F og tabell 2) [39]. Dette tyder på det sterkeste at metylering spiller en rolle i ekson regulation.Table en human genome og normal prøve informasjonen lende og intergeniske omegn
human Genome Informasjon
Normal tilleggsinformasjon
Relativ Enrichment Ratio
Funksjonell Kategori
Lengde (bp)
ratio product: # av CpG
ratio
Leser
ratio
vs. lengde
vs. CpG Count
Genic
1,184,139,094
39.46
13,262,253
47.09
20,854,434
53.25
1.35
1.13
Exon
68,035,894
2.27
1,808,089
6.42
4,350,405
11.11
4.90
1.73
Intron
1,122,817,725
37.41
11,613,113
41.23
17,358,273
44.32
1.18
1.07
Intergenic
1,816,976,186
60.54
14,901,610
52.91
18,310,273
46.75
0.77
0.88
Human Genome
3001115280
100
28163863
100
39164707
100
1.00
1.00
Tabell 2 menneskelige genom og normal prøve opplysninger av genet kommenterte regioner
human Genome Informasjon
Normal tilleggsinformasjon
Relativ Enrichment Ratio <.no> Funksjonell Kategori
Lengde (bp)
Ratio product: # av CpG
Ratio
Leser
Ratio
vs. lengde
vs. CpG Count
Upstream en kb
24468069
0,82
937748
3.33
535593
1,37
1,68
0,41
5'UTR Eksoner
8.436.529
0,28
411563
1,46
292654
0,75
2,66
0,51
Kode Eksoner
33384619
1.11
1.077.913
3,83
3.448.755
8,81
7,92
2.30
3'UTR Eksoner
28387978
0,95
378012
1,34
806832
2,06
2.18
1.53
Nedstrøms 1 kb
23136263
0,77
340866
1.21
551071
1,41
1.83
1.16
Menneskelig genom
3001115280
100
28163863
100
39164707
100
1.00
1.00
endringer i DNA-metylering mønstre forbundet med magekreft
Når gjennomsnittlig kromosom MES av kreft methylome ble sammenlignet med kontroll vev, fant vi at alle kromosomene i kreftvev tendens til å bli hypomethylated (tilleggsfiler 2: Figur S5). Med kromosom-wide utsikt, ble CGI-rike regioner funnet å være spesielt hypermethylated, mens gjenta rike regioner ble mye hypomethylated (figur 2A, ytterligere fil 2: Figur S6). Å analysere metylering endringer i genomiske elementer, justert vi hvert element på start- og slutt områder og deretter fått den gjennomsnittlige MES ved hver respektive posisjoner. Påfallende, vi har oppdaget hypermethylation i oppstrømsområdet, spesielt fra 500 bp oppstrøms for transkripsjonsstartsetet (figur 2B). Dette er i samsvar med hypermethylation av promotorområdene hyppig observert i kreft. Figur 2 Sammenligning av metylering mønstre i normal og kreftvev. (A) Gjennomsnittlig MES kurve for normal (svart) og kreft (rød) vev i kromosom 19 (til venstre). De gjennomsnittlige MES for CGIer, Gene organer, og gjentar (til høyre). (B) DNA metylering av gen kommentert elementer. Hvert element (oppstrøms en kb, exon, intron, og nedstrøms en kb) ble delt inn i 20 binger og gjennomsnittlig MES ble oppnådd for hver hylle av alle tilsvarende elementer. (C) DNA metylering ved avskrift ender og koding regionen ender. De gjennomsnittlige MES ble oppnådd i et glidende 50-bp vindu i henhold til dens avstand fra den transkript start (første) og enden (andre) for CGI-positive aktivatorer, så vel som transkriptet start (tredje) og utgangen (fjerde) for CGI- positive arrangører. (D) DNA-metylering av totalt 5 'UTR eksoner (til venstre) og 5' UTR kodende eksoner (høyre).
Område sentrert ved transkripsjonsstartsetet viste helt forskjellige mønstre avhengig av tilstedeværelsen av et CGI, noe som reflekterer den lave metylering status for CGI-holdige arrangører (Figur 2C). Vi fant også at i kreftvev, bemerkelsesverdig hypermethylation av CGI-holdige arrangører oppstår og at tettheten av CPGs er avgjørende for økningen i DNA metylering (figur 2C). For ytterligere å analysere hvorvidt 5 'regioner av genene ble hypermethylated på samme måte som genet promotorer, sjekket vi metylering mønsteret av de første eksoner. Interessant, fant vi at den første ekson ble hypermethylated bare når det var 5'-enden av en koding ekson, men ikke når det var en 5 'UTR ekson (figur 2D). Disse regionene inneholdt også høy CpG tetthet. Derfor CGIer på oppstrøms områder av gener, arrangøren og koding start synes å være de viktigste målene for DNA hypermethylation i kreft.
Metylering mønster av CpG øyer
å utforske sammenhengen mellom plasseringen av CGIer og DNA-metylering, subgrouped vi CGIer i henhold til deres posisjon i genomet. Nærmere bestemt ble de kategorisert som 5 '(som ligger mellom 1 kb oppstrøms og den kodende startsetet av et gen), intragenic (intragenic CGIer utenfor den 5' ende), og intergeniske (lokalisert i ikke-kallende region) (Tilleggs fil 1: Tabell S5). Selv CpG tetthet var lik blant de tre gruppene, non-5 'CGIer (intragenic og intergeniske CGIer) var signifikant mer denaturert enn 5' CGIer (tilleggsfiler 2: Figur S7). Vi ytterligere sammenlignet den gjennomsnittlige MES av subgrouped CGIer og fant metylering av alle CGIer var generelt økt. Men den relative differensial MES foreslått at endringen i metylering i 5 'CGIer var betydelig større enn for andre CGIer (figur 3A), som gjenspeiler de viktige roller av 5' CGIer i kreft. Omfanget av 5'CGI hypermethylation signifikant korrelert med overlapping av transkripsjonsstartsetet (figur 3B). Figur 3 DNA metylering av CpG øyer. (A) Relative differensial MES av subgrouped CGIer. (B) Sammenheng mellom differensial CGI metylering og avstand til transkripsjonsstartsetet. (C) Sammenheng mellom genekspresjon nivå og den hypermethylation av CGIer. (D) Metylering-spesifikk PCR fra histpngenene som viser de høyeste differanse MES verdier. . M1 og U1 tilsvarer HIST3H2A, mens M2 og U2 tilsvarer HIST3H2B
For å utforske funksjonene i genene som gjennomgår differensial metylering ved 5 'CGIer, valgte vi gener med svært differensial CGI Mess (differensial MES > 1). Vi deretter utført genet ontologi (GO) analyse for å få innsikt i mekanismene som er ansvarlig i kreft (tabell 3). Når genene ble gruppert i ulike GO kategorier, fant vi at HOX
gensamlingene og nucleosome montering relaterte gensamlingene var mål for hypermethylation, mens apoptose-relatert gensamlingene var mål for hypometylering. Interessant, våre funn at HOX
gensamlingene var fortrinnsrett mål for DNA metylering er konsistent med en tidligere rapport [40]. I tillegg genet tomter bekreftet at hypermethylation var CGI-spesifikk i kreft (tilleggsfiler 2: Figur S8). For å estimere endringer i uttrykk mønstre forårsaket av hypermethylation av 5 'CGIer, utførte vi en funksjonell analyse av genuttrykk data fra cDNA microarray eksperimenter. Hypermethylation av 5 'CGIer var signifikant korrelert med nedregulering av gener (p
= 0,03) (figur 3C, tilleggsfiler 3: Tabell S6 og S7). Dette indikerer at lyddemping av gener ved metylering kan bli direkte berørt av graden av CpG tetthet og 5 'CGI hypermethylation. Vi analyserte DNA metylering status av gener med hypermethylated 5 'CGIer og downregulated uttrykk mønstre. Blant disse var det genet som koder for histon H2B type 3-B (HIST3H2BB
). Analyse av HIST3H2BB
arrangøren metylering ved hjelp av metylering spesifikke PCR viste at de fleste kreftpasienter (8/10, 80%) viste økt metylering i promotorområdet (Figur 3D) .table 3 Funksjonell annotering gruppering av gener med hypermethylated 5'CGIs
Stempler Cluster en
Enrichment Score: 3,27
Count
P_VALUE
GOTERM_BP_FAT
nucleosome montering
11
3.90E-04
GOTERM_BP_FAT
kromatin montering
11
5.20E-04
GOTERM_BP_FAT
protein-DNA kompleks sammenstilling
11
7.40E-04
Stempler Cluster 2
Enrichment Score: 2,92
Count
P_VALUE
Interpro
Histone kjerne
8
6.80E-04
SP_PIR_KEYWORDS
nucleosome kjerne
8
8.60E-04
GOTERM_CC_FAT
nucleosome
8
3.10E-03
Stempler Cluster 3
Enrichment Score: site
17
5.10E-03
INTERPRO
Homeobox
17
5.70E-03
SP_PIR_KEYWORDS
Homeobox
17
5.80E-03
INTERPRO
Homeodomain-related
17
6.40E-03
SMART
HOX
17
1.40E-02
D4
3
9.10E-03
SP_PIR_KEYWORDS
embryo
3
3.30E-02
PIR_SUPERFAMILY
PIRSF002612:homeotic
 Lekk tarm og romfart - mekanismen avslørt
Lekk tarm og romfart - mekanismen avslørt
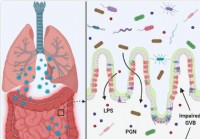 Utett tarm og mikrobiell dysbiose kan bidra til cytokinstorm i alvorlig syke COVID-19-tilfeller
Utett tarm og mikrobiell dysbiose kan bidra til cytokinstorm i alvorlig syke COVID-19-tilfeller
 Mikrober kan forutsi dødelige utfall hos ventilerte COVID-19-pasienter
Mikrober kan forutsi dødelige utfall hos ventilerte COVID-19-pasienter
 Tilstedeværelse av visse tarmbakterier hos mødre kan beskytte babyer mot matallergi
Tilstedeværelse av visse tarmbakterier hos mødre kan beskytte babyer mot matallergi
 GSK-3-hemmere viser løfte om behandling av koronavirusinfeksjoner
GSK-3-hemmere viser løfte om behandling av koronavirusinfeksjoner
 Xylitol og grapefruktfrø ekstrakt viser løfte om å forhindre SARS-CoV-2 infeksjon,
Xylitol og grapefruktfrø ekstrakt viser løfte om å forhindre SARS-CoV-2 infeksjon,
 Kryptosporidiose forverret av ofte brukt probiotika
Forskere har funnet ut at infeksjonen med tarmparasitten Cryptosporidium parvum forverres hos mus som har fått et probiotikum. Bildekreditt:Alpha Tauri 3D Graphics / Shutterstock Som
Kryptosporidiose forverret av ofte brukt probiotika
Forskere har funnet ut at infeksjonen med tarmparasitten Cryptosporidium parvum forverres hos mus som har fått et probiotikum. Bildekreditt:Alpha Tauri 3D Graphics / Shutterstock Som
 Edderkoppespeptid kan hjelpe til med å stoppe smerter ved irritabel tarm
Forskere fra University of Queensland har funnet ut at et spesifikt peptid som finnes i edderkoppgift kan ha egenskaper som kan gjøre det nyttig for å lindre smerter hos pasienter med irritabel tarm.
Edderkoppespeptid kan hjelpe til med å stoppe smerter ved irritabel tarm
Forskere fra University of Queensland har funnet ut at et spesifikt peptid som finnes i edderkoppgift kan ha egenskaper som kan gjøre det nyttig for å lindre smerter hos pasienter med irritabel tarm.
 Å håndtere cøliaki
Cøliaki er en immunreaksjon mot å spise gluten. Gluten er et protein som finnes i hvete, rug, bygg, og triticale. De fleste tenker på glutens med pasta, brød, pizza skorpe, o.l, men det er også i noen
Å håndtere cøliaki
Cøliaki er en immunreaksjon mot å spise gluten. Gluten er et protein som finnes i hvete, rug, bygg, og triticale. De fleste tenker på glutens med pasta, brød, pizza skorpe, o.l, men det er også i noen