Estratto
Sfondo
Riduzione delle cellule tumorali solide rappresenta un ostacolo fondamentale in oncologia clinica. Quindi, la caratterizzazione molecolare dei percorsi che regolano chemiosensibilità è un prerequisito fondamentale per migliorare la terapia del cancro. Il fattore HIF-1α ipossia-inducibile è stata collegata a chemiosensibilità mentre i meccanismi molecolari alla base rimangono in gran parte sfuggente. Pertanto, abbiamo ampiamente analizzato il ruolo di HIF-1α nel determinare chemiosensibilità concentrandosi sulle vie molecolari responsabili.
RNA interferenza è stata applicata per inattivare HIF-1α o p53 nel cancro gastrico umano linee di cellule AGS e MKN28. Gli agenti chemioterapici 5-fluorouracile e cisplatino sono stati utilizzati e chemiosensibilità è stata valutata mediante saggi di proliferazione cellulare e la determinazione della distribuzione del ciclo cellulare e l'apoptosi. L'espressione di proteine p53 e p53 bersaglio è stata analizzata mediante Western Blot. l'attività di NF-kB è stato caratterizzato per mezzo della determinazione spostamento mobilità elettroforetica. L'inattivazione di HIF-1α in cellule di cancro gastrico ha provocato robusta elevazione della chemiosensibilità. Di conseguenza, le cellule HIF-1α-competenti mostrato una riduzione significativa della senescenza indotta da chemioterapia e apoptosi. Sorprendentemente, questo fenotipo era completamente assente in p53 In sintesi, abbiamo identificato HIF-1α come un potente regolatore di attività di p53 e NF-kB in condizioni di stress genotossico. Concludiamo che p53 Visto:. Rohwer N, C Dame, Haugstetter A, B Wiedenmann , Detjen K, Schmitt CA, et al. (2010) ipossia-inducibile fattore 1α Determina cancro gastrico chemiosensibilità attraverso la modulazione di p53 e NF-kB. PLoS ONE 5 (8): e12038. doi: 10.1371 /journal.pone.0012038 Editor: Deb Fox, L'Istituto di ricerca per i bambini all'ospedale dei bambini di New Orleans, Stati Uniti d'America Ricevuto: 28 Aprile 2010; Accettato: 19 Luglio, 2010; Pubblicato: 10 Agosto 2010 Copyright: © 2010 Rohwer et al. Questo è un articolo ad accesso libero distribuito sotto i termini della Creative Commons Attribution License, che permette l'uso senza restrizioni, la distribuzione e la riproduzione con qualsiasi mezzo, a condizione che l'autore originale e la fonte sono accreditati Finanziamento:. Questo studio è stato sostenuto da sovvenzioni dal Deutsche Forschungsgemeinschaft (http://www.dfg.de) per TC (CR 133 /2-1, 133 /2-2 e 133 /2-3), e NR (Graduiertenkolleg 276/4 - "Signalerkennung und -umsetzung"). TC è stata sostenuta anche da una sovvenzione da parte del Berliner Krebsgesellschaft e.V. (Http://www.berliner-krebsgesellschaft.de, CRFF200804). I finanziatori avevano alcun ruolo nel disegno dello studio, la raccolta e l'analisi dei dati, la decisione di pubblicare, o preparazione del manoscritto Competere interessi:.. Gli autori hanno dichiarato che non esistono interessi in competizione Introduzione intrinseca e acquisito resistenza ai farmaci sono le cause primarie di limitata efficacia della chemioterapia nella maggior parte dei tumori maligni gastrointestinali, tra cui il cancro gastrico [1], [2]. La resistenza ai farmaci rappresenta un fenomeno complesso e multifattoriale relative al microambiente tumorale, ad esempio ipossia, acidosi e infiammazione così come la stessa cellula neoplastica [3]. resistenza cellulare può essere inerente allo specifico background genetico della cellula tumorale o il risultato di mutazioni e alterazioni epigenetiche dopo la terapia antiproliferativo [4], [5]. Il fattore di trascrizione fattore ipossia-inducibile 1 (HIF-1 ) costituisce un regolatore chiave di adattamento cellulare all'ipossia ed è stato coinvolto nella resistenza ai farmaci [6] - [8]. La proteina HIF-1 è un eterodimero composto di un β-subunità costitutivamente espresso (ARNT (arilico recettore translocator nucleare)) e una α-subunità ipossia-inducibile [9]. In condizioni normossiche, l'attività di HIF-1α può essere indotta da vari fattori di crescita, citochine, oncogeni attivati o perdita-di-funzione mutazione geni oncosoppressori [10]. HIF-1α è centralmente coinvolto in molteplici aspetti della tumorigenesi, tra cui la proliferazione delle cellule tumorali, l'angiogenesi, metastasi, così come la risposta alla chemioterapia e la radioterapia [11]. HIF-1α è sovraespresso in un vasto numero di tumori solidi, e l'espressione tumorale HIF-1α è spesso associata a prognosi infausta [12] - [15]. Inoltre, l'inibizione di HIF-1α mediante RNA interference o composti farmacologici ha dimostrato l'efficacia antitumorale in vari modelli murini di cancro [16]. Un contributo di HIF-1α per chemoresistance di cellule neoplastiche è stata osservata in un ampio spettro di tumori solidi, tra cui il cancro gastrico [6] - [8], [17] - [20]. Tuttavia, i meccanismi molecolari alla base, nonché il ruolo di HIF-1α per la resistenza ai farmaci in condizioni normossiche rimangono in gran parte sfuggente [8], [18], [21]. Qui, noi identifichiamo la soppressione di p53 e la promozione di attività di fattore nucleare kB (NF-kB) come meccanismi centrali per di HIF-1α sensibilità che ne determinano ruolo contro il 5-fluorouracile (5-FU) e cisplatino in cellule di cancro gastrico umano. Risultati HIF-1α determina sensibilità delle cellule di cancro gastrico verso gli agenti chemioterapici 5-fU e cisplatino inattivazione funzionale di HIF-1α è stato raggiunto da trasduzione lentivirale di AGS e cellule MKN28 con piccoli RNA interferenti (siRNA) mira specificamente HIF-1α. Questo approccio sperimentale prodotto un atterramento altamente efficiente dimostrato da una quasi totale fallimento di cellule trasdotte per indurre proteina HIF-1α in risposta all'ipossia come pubblicato in precedenza [22]. Per valutare l'importanza di HIF-1α per la sensibilità delle cellule di cancro gastrico umano verso agenti chemioterapici stabiliti, abbiamo confrontato gli effetti di 5-FU e cisplatino in HIF-1α-competenti (strapazzate, "SCR") e HIF-1α-carenti (atterramento, "KD") le cellule AGS. inattivazione funzionale di HIF-1α spostato la dipendenza dose di inibizione della crescita verso concentrazioni di farmaco inferiori (Figura 1A e Figura S1), suggerendo che HIF-1α è in grado di ridurre la chemioterapia suscettibilità delle cellule di cancro gastrico in condizioni normossiche. In linea con le precedenti relazioni [6] - [8], [17], [18], l'esposizione all'ipossia maggiore resistenza al 5-FU in cellule AGS, tuttavia l'inattivazione di HIF-1α portato robusto aumento della chemiosensibilità in condizioni di ipossia ( Figura S2). In un approccio complementare, abbiamo studiato le conseguenze di iperespressione di HIF-1α (pcDNA HIF-1α) per la chemiosensibilità delle cellule AGS. cellule AGS overexpressing HIF-1α erano notevolmente più resistenti al trattamento con 5-FU (Figura 1B). Stabile l'espressione di HIF-1α è stata confermata da HRE (ipossia reattivo elemento) saggio luciferasi giornalista (Figura 1C). Questi risultati suggeriscono fortemente che HIF-1α limita l'azione citotossica 5-FU e cisplatino in cellule di cancro gastrico umano e che l'inattivazione di HIF-1α potrebbe avere effetti benefici sulla chemiosensibilità. Abbiamo iniziato una caratterizzazione della inibizione della crescita osservata analizzando i modelli di distribuzione del ciclo cellulare dopo la chemioterapia. G 1-sincronizzato, le cellule AGS siero-fame sono stati rilasciati da G 0 /G 1 fase con l'aggiunta di profili di siero e del ciclo cellulare sono stati determinati dopo l'aggiunta di 5-FU. culture rilasciata di AGS non trattate prontamente progredito attraverso G 1 in S e G 2 /M fasi [22], mentre le cellule 5-FU-trattati sono rimasti in G 1 fase (non mostrato). È interessante notare che il mantenimento di 5-FU-dipendente delle cellule in G 1 fase è stata notevolmente aumentata in AGS KD rispetto alle cellule AGS SCR, in coerenza con G 1 cellule ciclo arresto (Figura 2A). Irreversibile arresto del ciclo cellulare è emerso come un modo importante dell'azione degli agenti antiproliferativi ed è caratterizzato da caratteristiche cellulari di senescenza [7], [23]. Di conseguenza, la frazione di cellule senescenti è stata determinata. Infatti, il trattamento con 5-FU ha portato ad una induzione robusto di senescenza in cellule AGS. Questa risposta è stata significativamente migliorata nelle cellule con perdita concomitante di HIF-1α (Figura 2B). Inoltre, l'induzione di apoptosi è stato suggerito da un aumentato 1 frazione pre G in istogrammi di DNA di cellule AGS KD 5-FU-trattati (non mostrato). Pertanto, l'analisi quantitativa della frazione cellulare per apoptosi è stata ottenuta sulla base della rilevazione di spaccati caspasi-3 (Figura 2C). In accordo con i dati sulla distribuzione del ciclo cellulare, la frazione apoptotica 5-FU-indotta è risultato significativamente aumentato nelle cellule AGS KD HIF-1α-carenti rispetto alle cellule HIF-1α-competente. indotta dalla chemioterapia senescenza e apoptosi entrambi sono stati intimamente legata al soppressore del tumore p53 per ottenere evidenze sperimentali per il ruolo proposto di p53 nella regolazione di HIF-1α-mediata di chemiosensibilità nelle cellule AGS, abbiamo p53 funzionalmente inattivato da RNA interference usando trasfezione transiente di anti-p53 siRNA (p53 si) o un controllo scrambled siRNA (SI SCR). P53 è stato efficacemente abbattuto, come indicato dal fallimento delle cellule trasfettate per indurre p21 effettori p53 e MDM2 in risposta a 5-FU trattamento (Figura 3A). È interessante notare che le cellule AGS KD trasfettate con p53 si erano significativamente meno suscettibili di inibizione della crescita del 5-FU rispetto alle cellule AGS KD trasfettate con siRNA di controllo (Figura 3B). In linea con questi risultati, G ritenzione ciclo 1 cellulare e l'apoptosi delle cellule AGS KD 5-FU-trattati sono stati ridotti di p53 atterramento in confronto alle cellule trasfettate con siRNA di controllo (Figura 3D e 3E). In netto contrasto, chemiosensibilità delle cellule AGS HIF-1α-abile, non è stata influenzata dalla inattivazione di p53 (Figura 3C). per confermare la regolazione di HIF-1α-dipendente di 5-FU risposta e per caratterizzare ulteriormente il contributo di p53, abbiamo esaminato una seconda gastrica linea di cellule di cancro umano (MKN28), che porta una mutazione missense nel> TP53 al codone 251. È interessante notare che, la cancellazione di HIF-1α nelle cellule MKN28 non è riuscito a migliorare la inibizione della crescita dopo l'esposizione a 5-FU (Figura 4A). Allo stesso modo, 5-FU-indotta G 1 accumulo e l'apoptosi delle cellule MKN28 non sono stati colpiti dalla perdita di HIF-1α (Figura 4B e 4C). In linea con questi risultati, i livelli di proteina di p53 e pRb sono rimasti invariati nelle cellule MKN28 5-FU-trattate per tutto il periodo di 24 ore, e l'induzione p21 era assente (Figura 4D). Tuttavia, quando la funzione di p53 è stata restaurata dal pretrattamento con il composto chimico PRIMA-1 [24] HIF-1α atterramento tradotto in un notevolmente migliorato 5-FU citotossicità (figura S3). Coerente con il ruolo stabilito di p53 in chemioterapia citotossica indotta /effetti citostatici, il trattamento con PRIMA-1 di per sé NF-kB è un importante mediatore del ruolo di HIF-1α in chemiosensibilità l'attivazione di NF-kB è associata con una protezione da apoptosi indotta da chemioterapia e, viceversa, l'inibizione della NF -κB può migliorare l'efficacia di agenti anti-neoplastici sia in vivo Per affrontare la significato funzionale di NF-kB per l'inibizione della crescita di 5-FU-indotta, abbiamo sovraespresso p65 (p65 pcDNA) nelle cellule AGS KD. Transfection di pcDNA p65, ma non il vettore di controllo vuoto, portato ad una significativa induzione di proteine p65 e l'attività trascrizionale NF-kB in cellule AGS KD (figura S4). Da segnalare, le cellule AGS KD HIF-1α-carenti sovraesprimono p65 erano considerevoli più resistenti al trattamento con 5-FU rispetto alle cellule AGS KD trasfettate con il vettore di controllo (Figura 5C), coerente con un ruolo essenziale di NF-kB nel mediare chemioresistenza nei confronti 5-FU in cellule di cancro gastrico. Nel loro insieme, l'attivazione simultanea di p53 e l'inibizione di NF-kB in 5-FU-trattati, è stata osservata cellule AGS HIF-1α-carenti. Per chiarire, se entrambi gli eventi sono interdipendenti, abbiamo studiato 5-FU-indotta attivazione di NF-kB in MKN28 cellule con mutante p53. Per chiarire il meccanismo sottostante molecolare superinduzione p53 in 5-FU trattati cellule HIF-1α-deficienti, abbiamo caratterizzato il ruolo delle specie reattive dell'ossigeno (ROS). ROS costituiscono un collegamento candidato (i) ROS sono potenti attivatori della funzione di p53 e fattori chiave presi in considerazione nella induzione di p53 da diversi agenti chemioterapici [28], e (ii) HIF-1α può sopprimere la generazione di ROS, diminuendo l'attività mitocondriale e biogenesi [22], [29], [30]. Di conseguenza, le cellule AGS KD sono stati pretrattati con la Ros-inibitori diphenyleneiodonium cloruro (DPI) o apocinina. Entrambi gli inibitori conferito una protezione significativa contro inibizione della crescita di 5-FU-indotta nelle cellule AGS KD (Figura 6A e 6B). Inoltre, DPI e apocinina quasi completamente impedito l'induzione di p53 e la sua valle p21 bersaglio in 5-FU-trattati cellule (Figura 6C e 6D). Questi risultati suggeriscono un incrocio di segnalazione HIF-1α con la risposta p53-mediata di 5-FU a livello di produzione di ROS. Per stabilire un ruolo causale di HIF-1α per il potenziale redox delle cellule AGS dopo il trattamento con 5-FU, livelli di ROS intracellulari sono stati determinati in cellule AGS KD e SCR mediante citometria a flusso. Abbiamo scoperto che i livelli di superossido intracellulari nelle cellule AGS KD 5-FU-trattati sono stati 2,5 volte superiori a quelli nelle cellule AGS SCR 5-FU-trattati (Figura S5), indicando che l'inattivazione funzionale di HIF-1α nelle cellule AGS ha provocato elevazione significativa e funzionale di stress ossidativo intracellulare anche sotto trattamento chemioterapico Discussione Il fattore di trascrizione HIF-1α è stato stabilito come importante mediatore della chemioresistenza ipossia-mediata [6] - [8]. , [17], [18], [20]. Qui, identifichiamo HIF-1α come un potente determinante di chemiosensibilità nelle cellule di cancro gastrico in condizioni normossiche. Applicando un sistema di siRNA lentivirus-based che buzz visualizzati notevolmente migliorato 5-FU e cisplatino tossicità nelle cellule di cancro gastrico HIF-1α-carenti. I dati disponibili sul ruolo di HIF-1α per la chemiosensibilità delle cellule tumorali in condizioni di normossia sono in conflitto. Mentre le cellule di fibrosarcoma HIF-1α-deficienti (HT1080) visualizzati in modo significativo una maggiore sensibilità verso etoposide in aria ambiente, il cancro del colon (HCT116) e epatoma (Hepa-1), le cellule non sono riusciti a farlo [6]. Unruh et al. controllo di progressione del cancro da chemioterapia si basa almeno in parte sulla induzione della senescenza cellulare. Recentemente, la perdita di HIF-1α ha dimostrato di causare senescenza precoce della immortalato fibroblasti embrionali murini in condizioni normossiche [32]. I nostri dati attuali suggeriscono che HIF-1α custodisce simile cellule di cancro gastrico contro senescenza indotta da chemioterapia in condizioni normossiche. Ciò costituisce il primo rapporto di elevato senescenza indotta da chemioterapia attraverso l'inattivazione funzionale di HIF-1α in una linea di cellule di cancro umano stabilito. Nelle cellule HIF-1α-carenti, abbiamo anche osservato migliorato induzione di apoptosi in risposta a 5-FU. Precedenti studi hanno riportato una riattivazione del fattore proapoptotica [6] Bid, o un cambiamento nella bilancia dei membri della famiglia Bcl-2 pro e antiapoptotiche per tenere conto di un aumento dei tassi di apoptosi seguenti inattivazione di HIF-1α in cellule di cancro gastrico trattati con farmaci [ ,,,0],. 18] il nostro studio identifica un nuovo meccanismo, per cui HIF-1α contrasta sia indotta da chemioterapia senescenza ed apoptosi: presentiamo la prova conclusiva per la capacità di HIF-1α per sopprimere l'induzione del tumore soppressore p53 in risposta a 5-FU in condizioni normossia. P53 è un cardine destino delle cellule determinante per il suo ruolo nella regolazione della progressione del ciclo cellulare e l'apoptosi in risposta allo stress cellulare e costituisce il gene più frequentemente mutato nei tumori umani [33]. Un'ampia varietà di agenti chemioterapici hanno mostrato per stabilizzare p53 e, viceversa, perdita di p53 costituisce un principio meccanismo di resistenza cancro verso chemioterapia [33], [34]. L'interazione di p53 e di HIF-1α è stato oggetto di dibattiti di lunga data come entrambe le relazioni positive e negative sono state pubblicate [35]. lavoro Tuttavia, l'intero precedentemente pubblicato concentrata sulla p53-HIF-1α-interazioni sotto ipossia (o anche anossico) condizioni. Per quanto a nostra conoscenza, i nostri esperimenti per la prima volta, forniscono la prova per la soppressione di p53 attività da HIF-1α in condizioni normossiche. Come conseguenza di p53 nelle cellule upregulation HIF-1α-deficienti, abbiamo osservato cambiamenti nel effettori a valle che sono collegati al irreversibile arresto del ciclo cellulare caratteristica della senescenza, per esempio stabilizzazione p21 e ipofosforilazione di pRb. Diverso da nostra osservazione, recente lavoro su chemioresistenza verso etoposide in HIF-1α-carente fibroblasti embrionali murine immortalati non ha rispettato una induzione di p21 [36]. Inoltre, HIF-1α stabilizzato p21 e p27, così come ha portato a ipofosforilazione di pRb durante la crescita arresto indotta dall'ipossia di immortalato fibroblasti embrionali murini e linfociti B della milza primaria [37]. Questi risultati contrastanti sono molto probabilmente spiegate dai tipi di cellule indagati: Mentre Goda et al Mentre p53 è stato più volte dimostrato di contrastare la funzione di NF-kB [38], [39 ], i nostri dati attuali indicano un ruolo per il soppressore del tumore nella regolazione dell'attivazione di NF-kB HIF-1α-dipendente. Oltre a p53, NF-kB è emerso come un secondo, determinante centrale della resistenza nei confronti degli agenti chemioterapici [40]. Diversi studi diversi hanno stabilito legami funzionali tra NF-kB e HIF-1α, anche se variabile posto HIF-1α a monte di NF-kB o viceversa. Per esempio, la stabilizzazione indotta dall'ipossia di HIF-1α nelle cellule muscolari lisce è sotto il controllo trascrizionale di NF-kB [41]. Allo stesso modo, i risultati ottenuti da topi knockout IKK-beta condizionali hanno confermato il ruolo centrale di NF-kB nel controllo della base e di espressione indotta dall'ipossia di HIF-1α in vivo In questo studio siamo stati in grado di individuare ROS come un punto di intersezione di HIF- 1α con la risposta allo stress cellulare, p53-mediata alla chemioterapia. Intracellulare ROS sono noti come potenti induttori di p53 e di partecipare alla attivazione di p53 da farmaci chemioterapici [45]. I mitocondri rappresentano la prima fonte di ROS intracellulari [46], e HIF-1α probabilmente contrasta la produzione di ROS a livello mitocondriale attraverso molteplici meccanismi tra cui l'inibizione della biogenesi mitocondriale e del piruvato spola nei mitocondri, riduzione dell'attività mitocondriale a causa di una maggiore utilizzazione della glicolisi e l'attivazione dell'autofagia mitocondriale [29], [30], [47], [48]. In precedenza, abbiamo stabilito un collegamento funzionale tra la riduzione HIF-1α-controllato di ROS e ancoraggio indipendenza di cellule di cancro gastrico [22], implicando HIF-1α nella patogenesi del cancro gastrico in assenza di ipossia. Ora scopriamo che il capacitiy di HIF-1α per limitare la produzione di ROS di cellule di cancro gastrico conferisce anche la resistenza agli agenti chemioterapici che funzionano attraverso l'attivazione di p53 (Figura 6E). È interessante notare che sono stati segnalati effetti crescenti sulla resistenza terapia attraverso la modulazione di p53 e ROS per HIF-2α [49]. Le isoforme HIF-alfa 1 e 2 mostrano un'ampia sovrapposizione di obiettivi HIF putativi e vincolante per gli elementi risposta ipossica e l'assegnazione definitiva degli effetti indotta da ipossia a uno isoforma non è sempre attuabile [50]. Bertout et al. In considerazione della necessità clinica di individuare i predittori di risposta per le opzioni di trattamento disponibili, il nostro risultati potrebbero trattamento decisioni potenzialmente diretti: da un lato, la conoscenza di HIF-1α sovraespressione potrebbe dirigere una scelta di farmaci che agiscono sostanzialmente in modo p53-indipendente. D'altra parte, un particolare beneficio può derivare dalla combinazione HIF-1-inibitori e agenti che danneggiano il DNA (ad esempio, 5-FU) nei tumori con p53 funzionale. Al contrario, una ridotta efficacia di HIF-1-inibitori potrebbe essere anticipata per il trattamento di tumori p53 difettoso, un aspetto che può costituire un fattore di confusione in studi clinici di HIF-1α-inibendo regimi di trattamento. coltura delle cellule e prodotti chimici AGS (CRL-1739, ATCC, Rockville, Maryland, USA) e MKN28 (JCRB Cell Bank, Tokyo, Giappone), le cellule sono state coltivate come colture monostrato in mezzo standard . Generazione di cellule AGS e MKN28 esprimono stabilmente sia siRNA specificatamente destinate HIF-1α (atterramento, "KD") o di controllo aspecifici siRNA (strapazzate, "SCR") è stato pubblicato in precedenza [22]. 5-fluorouracile (5-FU), cis-Diammineplatinum (II) dicloruro (cisplatino) ed il cloruro di superossido anione inibitori diphenyleneiodonium (DPI) e apocinina sono stati acquistati da Sigma-Aldrich (Germania) sciolti in DMSO. PRIMA-1 (per p53-riattivazione e l'induzione di una massiccia apoptosi) è stato ottenuto da Tocris Biosciences (Ellisville, Missouri, USA) e disciolto in acqua sterile. controllo del veicolo dei solventi è stato incluso in tutti gli esperimenti. La proliferazione cellulare saggio Per la determinazione della crescita cellulare, 3 × 10 4 cellule sono state seminate in triplicato in piastre da 24 pozzetti, ha permesso di collegare per 16 ore e poi trattati come indicato in normossia o condizioni di ipossia. Dopo il trattamento, le cellule sono state tripsinizzate, e le cellule vitali sono state contate con un emocitometro. Cell distribuzione del ciclo compreso il pre-G 1 frazione è stata determinata da istogrammi di DNA, come descritto [52]. L'apoptosi è stata quantificata anche dal rilevamento di attivi, spaccati caspasi-3 mediante citometria a flusso utilizzando un Alexa Fluor® anticorpi 488-coniugato (Cell Signaling Technology Danvers, Massachusetts, USA). attività di β-galattosidasi senescenza associata è stata valutata in preparazioni cytospin come descritto [53]. Immunoblot analisi lisati cellulari intere sono state preparate come descritto in precedenza [52], quindi deliberato un 10% sodio dodecil solfato gel di poliacrilamide e trasferito su nitrocellulosa (Amersham Biosciences, Freiburg, Germania). Macchie sono stati sondati con anticorpi contro p53 e CDK2 (Santa Cruz Biotechnology, Santa Cruz, California, Stati Uniti d'America), p21 (Oncogene Research Products, Bad Soden, Germania), ciclina A (Upstate, Temecula, California, Stati Uniti d'America), pRb (BD Pharmingen , Heidelberg, Germania), MDM2 (Calbiochem, San Diego, California, Stati Uniti d'America), p65 (Cell Signaling Technology) e actina (Sigma-Aldrich). anticorpi secondari sono stati coniugati con perossidasi di rafano (Dianova, Amburgo, Germania) e l'attività di perossidasi è stato visualizzato utilizzando il occidentale fulmine Chemiluminescenza reagente più (Perkin Elmer Life Sciences, Boston, Massachusetts, USA). quantitativa real-time PCR analisi Per real-time PCR, RNA cellulare totale è stato estratto con il reagente Trizol (Invitrogen, Karlsruhe, Germania). Prima filamento di cDNA è stato sintetizzato con un (dT) primer oligo e ™ First Strand Synthesis System SuperScript (Invitrogen). Quantitativa real-time PCR è stata eseguita utilizzando TaqMan PCR universale Mastermix (per β-actina) o SYBR Green PCR Master Mix (per A20 e cIAP1, Applied Biosystems, Darmstadt, Germania). sequenze primer sono forniti in Tabella S1. Per normalizzare la quantità di RNA di ingresso, reazioni di PCR sono state fatte con la sonda e primer per il β-actina. trasfezioni transienti di cellule AGS sono stati effettuati utilizzando Effectene Trasfezione Reattivo (Qiagen, Hilden, Germania) secondo il protocollo del produttore. Per gli studi di iperespressione, le cellule sono state seminate a 3 × 10 4 celle /24-bene e trasfettate con 100 ng di pcDNA HIF-1α (gentilmente fornito da Wanja Bernhardt, Universitätsklinikum Erlangen, Erlangen, Germania) o pcDNA p65 (gentilmente fornito da Hiroyasu Nakano, Jutendo University, Tokyo, Giappone), rispettivamente. Per HRE o saggio luciferasi di NF-kB, 3 × 10 4 celle /24 pozzetti sono stati co-trasfettate con 100 ng di Phre-Luc (un dono di Randall S. Johnson, UCSD, San Diego, California, Stati Uniti d'America) o IgκB-Luc (un dono di Florian R. Greten, Technische Universität München, Monaco di Baviera, Germania), e 30 ng di phRL-null (Promega, Mannheim, Germania). l'attività luciferasi è stata misurata con la luciferasi Reporter Assay sistema Dual (Promega), come descritto [54]. Per ottenere la soppressione p53 transitoria, le cellule sono state trasfettate AGS al 30% confluenza con 75 o 150 nmol /L si p53, ( Silenziatore estratti proteici nucleari sono state preparate come descritto [54]. EMSA è stata eseguita come descritto in precedenza [55] con 8 mg di proteina nucleare e 100 fmol /L di fine-radiomarcato 22 bp a doppio filamento di NF-kB consenso oligonucelotide (sezione in avanti: 5'-AGT TGA GGG GAC TTT CCC AGG C- 3 '; E3292, Promega). I campioni sono stati risolti mediante elettroforesi su un non-denaturazione gel di poliacrilammide al 5%.
cellule mutanti mentre l'inattivazione di p53 di per sé
non ha influenzato chemiosensibilità. HIF-1α notevolmente soppressa l'attivazione indotta da chemioterapia di p53 e p21 e la proteina retinoblastoma, eventualmente con conseguente arresto del ciclo cellulare. formazione ridotta di specie reattive dell'ossigeno nelle cellule HIF-1α-competenti è stato identificato come il meccanismo molecolare di inibizione di HIF-1α-mediata di p53. Inoltre, la perdita di HIF-1α abrogata, in maniera p53-dipendente, indotta da chemioterapia DNA-binding di NF-kB e l'espressione di anti-apoptotici geni target di NF-kB. Di conseguenza, la ricostituzione della subunità p65 NF-kB ha invertito la maggiore chemiosensibilità delle cellule HIF-1α-carenti.
Conclusione e significato
mutazioni in tumori umani tenere il potenziale per confondere l'efficacia di HIF-1-inibitori nella terapia del cancro
limiti di HIF-1α indotta da chemioterapia arresto del ciclo cellulare e l'apoptosi attraverso la soppressione di p53
. Così, abbiamo ipotizzato che p53 potrebbe contribuire alla citotossicità aumentata del 5-FU in caso di perdita di HIF-1α. Dopo il trattamento con 5-FU, proteina p53 gradualmente accumulate nelle cellule AGS, un effetto che è stato sorprendentemente migliorato nelle cellule AGS HIF-1α-deficienti (Figura 2D). Questa stabilizzazione di p53 è stata associata ad un aumento dei livelli di p21 inibitore ciclina-dipendente chinasi (CDK), un target trascrizionale ben consolidata e effettori a valle di p53 con le funzioni di arresto del ciclo cellulare, l'induzione senescenza e l'apoptosi (Figura 2D). Anche in questo caso, le cellule AGS HIF-1α-deficienti hanno mostrato un marcato forte aumento della p21 di cellule AGS HIF-1α-abile. Forte l'induzione di p21 si prevede di inibire l'attività di G 1 ciclina /CDK complessi, con conseguente ipofosforilazione di proteina del retinoblastoma (pRb) e il fallimento di indurre cyclins fase S, ad esempio ciclina A. Infatti, sia pRb ipofosforilazione e ridotti livelli di ciclina A sono stati confermati nelle cellule AGS KD 5-FU-trattati e - in misura minore - anche in AGS cellule SCR (Figura 2D). Questi cambiamenti confermano il mantenimento G 1 fase osservata in istogrammi di DNA e sono coerenti con l'irreversibile G 1 arresto osservato in senescenza indotta da chemioterapia. Così, i diversi risultati biologici di trattamento con 5-FU in cellule AGS HIF-1α-deficienti e -proficient nascono dalla regolazione differenziale di p53 e la sua valle p21 bersaglio.
L'inattivazione di p53 blunts il ruolo di HIF-1α per chemiosensibilità
HIF-1α non riesce ad influenzare chemiosensibilità in p53
cellule mutanti
leggermente ridotto la proliferazione delle cellule MKN28 e notevolmente migliorato l'efficacia di 5-FU in MKN28 cellule (Figura S3).
e in vitro
[25] - [27]. Pertanto, abbiamo determinato l'attività di NF-kB legame al DNA nelle cellule AGS HIF-1α-deficienti e -proficient dopo il trattamento con 5-FU mediante test di spostamento mobilità elettroforetica (EMSA). Il trattamento con 5-FU potentemente attiva NF-kB legame al DNA nelle cellule AGS SCR, con livelli di picco che si verificano 6 ore dopo l'esposizione a 5-FU (Figura 5A). Il trattamento con TNFa per 4 h servito come controllo positivo per l'attivazione di NF-kB. Inoltre, un supershift è stata indotta mediante un anticorpo anti-p65, confermando che il 5-FU indotte complessi NF-kB contenevano la subunità 65-kDa (p65). Perdita di HIF-1α attivazione inibito significativamente di NF-kB in risposta a 5-FU e TNF (Figura 5A). Coerentemente con questa osservazione, 5-FU trattamento anche non è riuscito a indurre i geni target di NF-kB cIAP1 e A20 nelle cellule AGS KD, mentre erano stati prontamente indotte nelle cellule AGS SCR (Figura 5B)
.
Entrambe 5-FU e TNF attivato NF-kB legame al DNA in un tempo- dipendente manner, indicando meccanismi p53-indipendenti di attivazione di NF-kB da 5-FU (Figura 5D). Tuttavia, diverso dal ritrovamento nelle cellule AGS, questa attivazione di NF-kB nel p53
linea cellulare mutante non è stato attenuato dalla inattivazione HIF-1α. Così, HIF-1α può sostenere l'attivazione di NF-kB indotta da chemioterapia contrastando meccanismi inibitori p53-dipendente.
formazione di ROS Altered è responsabile per la modifica HIF-1α-indotta di p53 attività
Riferito maggiore suscettibilità di HIF-1α-carente topo fibroblasti embrionali a carboplatino o etoposide sotto normossia nonché condizioni di ipossia [8]. Per quanto riguarda il cancro gastrico, una maggiore efficacia di 5-FU e vincristina è stata dimostrata in normossia in vitro
[18]. Bene in linea con i nostri risultati, entrambi gli studi hanno concluso un ruolo fondamentale per HIF-1α nel mediare chemioresistenza in condizioni normossiche. È interessante notare che un recente studio giapponese ha dimostrato minore efficacia della chemioterapia 5-FU a base di HIF-1α-esprimendo adenocarcinoma gastrico umani, rafforzando la percezione di HIF-1 come un fattore cruciale nella determinazione della chemioresistenza cancro gastrico [31].
. caratterizzato una risposta fisiologica all'ipossia in tipi di cellule non trasformate, abbiamo analizzato la risposta a gravi danni al DNA in linee cellulari di cancro stabiliti.
[42]. Al contrario, è stato dimostrato di essere controllato da HIF-1α nel contesto di apoptosi ipossia represso dei neutrofili [43] l'espressione del gene della subunità p65 NF-kB. La nostra scoperta di marcatamente ridotta attività di NF-kB in cellule HIF-1α-carente in seguito al trattamento con 5-FU è quindi perfettamente in linea con quest'ultimo rapporto. È interessante notare, abbiamo anche osservato significativamente ridotto legame della subunità NF-kB in cellule HIF-1α-carenti dopo stimolazione con TNF-alfa, un induttore consolidata di attività di NF-kB [44] DNA. Ciò solleva la questione pertinente in quali condizioni fisiologiche e fisiopatologiche HIF-1α è in grado di regolare l'attivazione di NF-kB. HIF-1α e NF-kB condividono importanza cruciale per vari processi come l'infiammazione, l'uccisione microbica e tumorigenesi. La natura molecolare esatta così come la gerarchia della loro interazione è più probabile cellulo e dipendente dal contesto e non può essere generalizzata.
Dimostrato che inibendo HIF-2α aumenta l'apoptosi indotta da radiazioni attraverso l'accumulo di ROS e il successivo aumento di p53 attività [49]. Inoltre, Roberts et al.
Ha mostrato che la resistenza contro la chemioterapia è parzialmente mediata dalla soppressione HIF-2α-mediata di p53 nelle cellule di carcinoma a cellule renali [51]. Quindi, le osservazioni riportate con la presente garantisce indagini sul potenziale ruolo di HIF-2α, un compito che è attualmente in corso nel nostro laboratorio.
Materiali e Metodi
Determinazione della distribuzione del ciclo cellulare e l'apoptosi mediante citometria di flusso
La quantificazione della senescenza
trasfezione transiente e giornalista luciferasi test
Selezionare siRNA, Applied Biosystems) e analizzati 48 ore dopo la trasfezione. Un non-specifici siRNA (SI SCR, Eurogentec, Seraing, Belgio) è stato utilizzato come controllo.
elettroforetica saggio mobility shift (EMSA)
 La disbiosi nel microbiota intestinale può causare una grave infezione secondaria nei pazienti COVID-19
La disbiosi nel microbiota intestinale può causare una grave infezione secondaria nei pazienti COVID-19
 Un tipo di batteri intestinali può aumentare il rischio di cancro intestinale
Un tipo di batteri intestinali può aumentare il rischio di cancro intestinale
 La luce ultravioletta B fa bene al microbioma intestinale
La luce ultravioletta B fa bene al microbioma intestinale
 I grandi fagi appena scoperti offuscano il confine tra vita e non vita
I grandi fagi appena scoperti offuscano il confine tra vita e non vita
 Le donne nate con taglio cesareo hanno un rischio maggiore di obesità e diabete
Le donne nate con taglio cesareo hanno un rischio maggiore di obesità e diabete
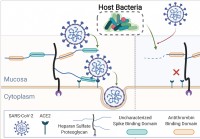 Il microbioma umano elimina i glicani della mucosa,
Il microbioma umano elimina i glicani della mucosa,
 Gravi complicanze del COVID-19 legate alla rottura della barriera intestinale
Uno dei principali obiettivi della ricerca nellattuale pandemia della malattia da coronavirus 2019 (COVID-19) è stata la necessità di comprendere i meccanismi operativi che causano le varie manifestaz
Gravi complicanze del COVID-19 legate alla rottura della barriera intestinale
Uno dei principali obiettivi della ricerca nellattuale pandemia della malattia da coronavirus 2019 (COVID-19) è stata la necessità di comprendere i meccanismi operativi che causano le varie manifestaz
 IBD molto più comune del previsto,
e non farà che aumentare in futuro Ci sono tre volte più persone con il disturbo intestinale cronico e debilitante chiamato malattia infiammatoria intestinale (IBD) di quanto si fosse mai pensato prim
IBD molto più comune del previsto,
e non farà che aumentare in futuro Ci sono tre volte più persone con il disturbo intestinale cronico e debilitante chiamato malattia infiammatoria intestinale (IBD) di quanto si fosse mai pensato prim
 I probiotici come terapia adiuvante per i pazienti COVID-19
Già nel 1892, Doderlein stabilì per primo lassociazione benefica dei microrganismi nel corpo umano. Perciò, sotto la pandemia di COVID-19, può essere utile valutare il ruolo dei microrganismi nel corp
I probiotici come terapia adiuvante per i pazienti COVID-19
Già nel 1892, Doderlein stabilì per primo lassociazione benefica dei microrganismi nel corpo umano. Perciò, sotto la pandemia di COVID-19, può essere utile valutare il ruolo dei microrganismi nel corp