Helicobacter pylori Citácia :. Rizzato C, Torres J, Plummer M, N Muñoz, Franceschi S, Camorlingo-Ponce M, et al. (2012) Kolísanie Helicobacter pylori cytotoxin asociovaných génov a ich vplyv na progresiu k rakoviny žalúdka: Dôsledky pre prevenciu. PLoS ONE 7 (1): e29605. doi: 10,1371 /journal.pone.0029605 Editor: Masaru Katoh, National Cancer Center, Japonsko Prijaté: 6. októbra 2011; Prijaté: 01.12.2011; Uverejnené: 03.01.2012 Copyright: © 2012 Rizzato et al. Toto je článok o otvorený prístup distribuovaný pod podmienkami Creative Commons Attribution licencie, ktorá umožňuje neobmedzené použitie, distribúciu a reprodukciu v nejakom médiu, za predpokladu, že pôvodný autor a zdroj sú pripísané Financovanie :. JT je príjemca exkluzivitu štipendia z Fundacion IMSS, Mexiko. Platcovia mal žiadnu úlohu v dizajne štúdie, zber a analýzu dát, rozhodnutie publikovať, alebo prípravu rukopisu Konkurenčné záujmy: .. Autori vyhlásili, že žiadne konkurenčné záujmy neexistujú Úvod Helicobacter pylori Najlepší marker virulencie vyznačujúci HP je cytotoxin spojené s gén patogenitu ostrov (cagPAI ), čo je 40 kb oblasť kódujúce chromozomálnej DNA približne 31 génov, ktoré tvoria typu IV sekrečnú systém (T4SS) sa premiestni bakteriálne produkty, do hostiteľskej bunky. ČAGA Mnoho z funkcií ČAGA sú umiestnené do iného C-terminálny tandemové usporiadaných opakujúce sa motív obsahujúci aminokyseliny Glu-Pro-Ile-Tyr-Ala (EPIYA motívy a, B, C a D). Kmene nesúce viac kópií západného typu EPIYA-C alebo východnej typu EPIYA-D sú navrhnuté, aby sa viac spájaný s rakovinou žalúdka a so zvýšeným ČAGA in vitro samotný stav ČAGA Prehlásenie etika Všetci účastníci podpísali informovaný písomný súhlas. Štúdia bola schválená etického preskúmania správnych radách inštitúcií zodpovedných za predmet náboru v každej z prijímacích stredísk. Na mexických vzoriek štúdia bola schválená etickými výbormi Instituto Mexicano del Seguro Social (IMSS) a General Hospital v Secretaria de Salud (SS), Mexico City, Mexiko. v prípade Venezuely vzoriek, etické vôľa k štúdiu bola získaná z Medzinárodná agentúra pre výskum rakoviny (IARC) etickou komisiou v Lyone, Francúzsko a Control Center Cancer v San Cristobal, Venezuela. populácii Venezuela. Použili sme 11 vzoriek DNA od žalúdočných biopsiou od subjektov postihnutých chronickou gastritídou bez atrofia prijatí do štúdie chemoprevence vo Venezuele [17], [18]. V študijné predmety (vek 35-69) V tejto štúdii sa regrutujú z účastníkov rakovina žalúdka riadiaci program Tachira štátu, ktorý bol založený na žalúdočné dvojitým kontrastným röntgene nasleduje gastroskopicky skúškou. Pacienti s niektorou rakoviny, vrátane rakoviny žalúdka, alebo iných závažných ochorení, ako je srdce, pľúc, obličiek alebo zlyhanie pečene a tehotných žien neboli oprávnené. Sedem žalúdočné biopsie boli odobraté z preddefinovaných miest, päť pre histologické vyhodnotenie a dva boli zmrazené na H pylori 84 vzoriek bolo od pacientov navštevujúcich Gastroenterology jednotka General Hospital Mexiko (Secretaría de Salud) a nemocnice onkológia ( Instituto Mexicano del Seguro Social), obaja nemocnice v Mexico City. Tridsať päť pacientov bolo postihnutých chronickou gastritídou a 49 s rakovinou žalúdka. Pacienti boli starší ako 30 rokov, ktorý bol konzultovaný pretože gastroduodenálnych symptómov (General Hospital), alebo v dôsledku pravdepodobného rakovinou žalúdka (onkológie FN), a boli naprogramované pre endoskopiu a biopsia na diagnostické účely. Jedinci, ktorí predtým liečení rakoviny, boli na antibiotiká, liečba anti-HP alebo nesteroidné protizápalové lieky dva týždne pred začiatkom štúdie, alebo mal iné závažné chronické choroby boli vylúčené. Žalúdočné biopsie boli umiestnené v sterilnom 0,9% roztoku fyziologického roztoku, homogenizované a zaočkovaný krvným agarom bázy (BBL, MD) doštičky doplnené 5% ovčej krvi za HP kultúry. Doštičky boli inkubované pri teplote 37 ° C v 9% CO 2 atmosfére po dobu až 5 dní. HP bol identifikovaný kolónií a mikroskopické morfológiu a pozitívne oxidázy, katalázy, a ureasové testy. Z každej primárnej rast, 7 až 10 jednotlivých kolónií každej boli izolované z antra a korpusu a propagované na krvnom agare médiu. V tejto štúdii sme analyzovali 43 vzoriek z kultivovaných kmeňov a 41 priamo zo zmrazených biopsiou. Hlavné charakteristiky populácie sú uvedené v tabuľke 1. DNA extrakcie pre venezuelské a mexickej vzorky biopsia bola DNA extrahovaná zo zmrazených tkanív s použitím QIAamp DNA Micro Kit (Qiagen, Hilden, Nemecko) podľa inštrukcií výrobcu. Pre kultivovaných kmeňov DNA sa čistí pomocou EDTA thiokyanidu-Sarkosylu metódu guanidínu (GES) [19]. Použili sme zarovnanie HP sekvencií z verejných databáz pre identifikáciu sekvencií vhodných pre návrh PCR primérov. obmedzený sme naše vyhľadávacie databázy do západných kmeňov HP, ktoré sú viac pravdepodobné, že bude podobné kmene nájdené v našej štúdii vzoriek. Navrhli sme dláždené amplikóny s veľkosťou od 312 do 876 bp. Priemerná veľkosť sekvencie číta s 454 sekvenčná technológia je 450 bp, teda dopredu číta a vzad číta prekrytie aspoň čiastočne, čím sa zlepší spoľahlivosť výstupu. Päť ČAGA Ďalej sme použili, as referenčná, tri kmene 26695 (NC_000195), J99 (NC_000921) a G27 (NC_0011333), ktorých genómy boli kompletne sekvenované [22], [23], [24]. 454 sekvenovania potom, čo boli optimalizované podmienky PCR sme resynthesized rovnaké priméry používané na PCR s multiplexu tagov (identifikovania sekvencie z každého konkrétneho vzorky) a adaptérov, a zosilnený cieľových regiónoch pomocou DNA zo vzoriek. Druhá PCR bola vykonaná za použitia označených primerov, za účelom zvýšenia množstva materiálu. Všetky PCR amplimers boli potom čistené, kvantifikovaná spektrofotometricky a sústredené v ekvimolárnej množstvách. Knižnica generácie pre 454 FLX sekvencovanie bolo vykonané pomocou štandardných protokolov výrobcu (454 Life Sciences Corporation, Branford, CT, USA). Stručne povedané, adaptéry výrobca potrebné pre spracovanie a sekvencovanie boli pridané do konečných každého zoskupenia označených produktov PCR ligácia. Jednotlivé molekuly PCR produktov nesúcich správne adaptéry boli hybridizovány na jednotlivé guličky, klonovo zosilnené v následnom emulzie PCR a každý zásobník nanesie na 1/16 v picotiterplate pre sekvencovania pomocou technológie 454 GS FLX Titanium. Po spracovaní a základným Volanie pomocou proprietárny softvér výrobcu (454 Life Sciences Corporation, Branford, CT, USA, Software verzia 2.0.00 10. 2008) Výsledná číta boli triedené podľa dopredu začlenený šiestich základných značiek. Genomické analýzy sekvencie od 454 technológií bolo vykonané za nich HP izoláty s > 200krát priemernej pokrytie (minimálne 59x, 580x maximum). Výsledné contigs boli zostavené s použitím génovej sekvencie HP kmeňa 26695 [23] ako lešenie. Nemáme pozorované značné rozdiely v kvalite výstupu medzi DNA z kultivovaných kmeňov a DNA z biopsiou. Aby bolo možné posúdiť kontrolu kvality dát sme porovnávali 454 sekvenčného spracovania dát z referenčného kmeňa 26695 a publikovanú sekvencií v databáze NCBI (NC_000915); zhoda bola v priebehu viac ako 99%. Tiež sme sekvenuje 9 venezuelskej vzorky s tradičným spôsobom Sangerovo sekvenovania, pozorovanie konkordancii >. 99% medzi metódami Sangerovo sekvenovania ČAGA Suroviny sekvencie boli analyzované automaticky so softvérom 454, a kvalitné výsledky boli pridelené. Výsledná sekvenčný výstup, ako z 454 vo formáte SFF a Sanger v abi formáte, bola analyzovaná s viacnásobné sekvenčné zoradenie softvér (napr softvérovú platformu Geneious: http://www.geneious.com/~~HEAD=dobj), ktorý číta zostavený všetky patriace k rovnaký vzorka, potom sekvencie všetky vzorky boli uvedené do súladu s referenčnou sekvencií a jednonukleotidových polymorfizmov, ako aj malých inzercie a delécie boli identifikované. Aby sa predišlo prípadným artefaktov z sekvenovanie a obmedziť varianty s klinicky a štatisticky zmysluplné frekvencie, sme vybrali varianty s jednoznačným volania pozorovaných v aspoň 10% synonymické (N = 175) a 20% pre nonsynonymous (N = 206) varianty pôvodne testované vzorky a referenčné kmene (celkom 381). SAS verzia 9.2 bola použitá na odhad Logitové miery pravdepodobnosti (OR) a 95% interval spoľahlivosti (CI) pre rakovinu žalúdka súvisiace s každou variant, a vypočítanie p -hodnoty na rozdiely vo variante frekvenciách medzi rakovinou žalúdka a gastritídou podľa Fisherovho exaktného testu (2-sided). Bonferroniho korekcia bola použitá pre výpočet hodnoty p upravená s prihliadnutím na viacnásobná porovnávanie vydelením surovej hodnoty p s 381. Genetická variabilita v siedmich HP cagPAI génov Súhrn genetickej variability zistených v sedem génov je uvedená v tabuľke 2. ako sa dalo očakávať, sme pozorovali vysoký stupeň variability (vypočítaná ako počet miest ukazuje variant von z celkového počtu miest v géne), obaja na úrovni kyseliny DNA a aminokyselín. Variabilita nukleotidu sa pohybovala od 8,03% do cagV porovnávali sme frekvencia 381 vybraných genetických variantov medzi rakovinou žalúdka a chronických prípadov gastritídy. Potom sme zistili, non-synonymom (tabuľka 3) a synonymické (tabuľka 4) varianty, ktoré ukázali výrazné rozdiely medzi gastritídy a onkologických prípadov, ktoré spĺňajú jednu z nasledujúcich kritérií: (1) absolútna varianta frekvencie medzi gastritídy a rakovinou skupiny sa líšili najmenej 25%; (2) frekvencia variantov u karcinómu žalúdka aspoň dvakrát vyššia ako v žalúdku, a (3) variant frekvencie v žalúdku aspoň dvakrát vyššia ako u karcinómu žalúdka. Dvadsať päť SNP (11 non-synonymá a 14 synonymické) dosiahla štatisticky významné rozdiely (p menšie ako 0,05, obrázok 1), ktorý sa nachádza v časti ČAGA C-koncovej oblasti (polohy 2670 do 3100), bola vysoko variabilné v klinických izolátov podľa vzoru motívov EPIYA (obrázok 2). Pozorovali sme 524 polymorfné miest, z ktorých sme analyzovali 148 zvolená kritériá predtým popísaných (kompletného katalógu ČAGA Analýza regiónu EPIYA potvrdila, že všetky sekvencie boli západného typu ČAGA, tj, ABC (82%), ABCC (13%), ABABC (3%), AABCC (1%) a ABCCC (1%). Sme nespozorovali rôzneho rozdelenia týchto typov ČAGA medzi rakovinou a gastritídy prípadoch (p = 0,2342), Podrobné výsledky sú uvedené v tabuľke 5 a obrázku 2. Sledovali sme 3 varianty EPIYA motívu: jedna vzorka mal gastritída EPIYV An motív, 50% z motívov B ukázala variant EPIYT (žiadny štatistický inú distribúciu v rakoviny a gastritídu prípade) a jeden C motívom prípadu rakoviny ukázal variant EPLYA. výsledky v druhej CAG Genetická variabilita cagC klietke V cagC Cage varianta C1039T, pri analýze ako jediná zmena, predpovedá aminokyselinovú zmenu lyzínu na phenilalanine (zmena kodónu CTT do TTT), zatiaľ čo SNP T1041G leží na tretej pozícii toho istého kodóne, a v prípade, analyzovať jediná zmena sa predpokladá synonymické varianty (codon zmeniť CTT na CTG). Avšak vo všetkých vzorkách, ktoré sme analyzovali dve variantné alely boli pozorované spoločne, a preto sme pozorovali iba dve varianty kodónov (CTT TTG), a ktoré kódujú rovnakú aminokyselinu, lyzín. T1905C polymorfizmus je synonymom pre variant v tretej polohe kodónu (GTT GTC variant kodóne), ktorá kóduje valín. V CAGL cagT V CAG Gamma Diskusia Od svojho objavu v roku 1996 [25], bude cagPAI, ktorý skrýva virulentné gény HP, bol pravdepodobne najviac intenzívne študoval časť genómu HP. IV sekrečnú systém typu kóduje proteíny, ktoré tvoria ihlicovité štruktúry spájajúcej HP do cytoplazmy epitelové bunky žalúdočnej vstrekovať onkogénne proteín ČAGA a peptidoglykanov. Komponenty tejto štruktúry zahŕňajú a) Pilus zložky, CagC (homológy Agrobacterium tumefaciens Zistili sme, minimálne rozdiely, a to ako v nukleotidu a aminokyselín úrovni vo vnútorných komponentov jadra T4SS (CagT , CagV, a klietka, 5,7%, 5,9% a 5,9% aminokyselín variácie, v tomto poradí), a najväčšia zmena v exponovaných častí: integrín viažuci proteín CAGL, extracelulárnej pilus hlavnou zložkou CagC a vylučovaný proteín ČAGA (12,2%, 23,5% a 31%, aminokyseliny variácie, v tomto poradí). Tieto výsledky potvrdzujú, že genetická variabilita v súčastiach cagPAI je ovplyvnená predovšetkým ich lokalizácia v T4SS, s vyššou variability v proteínoch exponovaných v bakteriálnom povrchu, možno ako odpoveď na imunologické tlaku. Je zaujímavé, CAG Gamma bola výnimka (19,5% aminokyselina variácie), tento proteín bol navrhnutý na pobyt v periplazmě HP, na ktoré pôsobia ako peptidoglykánu hydrolázy, prepichnutím vonkajšej membránu HP, a tým pomáha vystaviť T4SS Pilus na externé médiá [28]. Je možné, že CAG Gamma plní túto funkciu aj ako štruktúrne súčasť exponované Pilus, kde by sa tiež mohla pôsobiť cez membránu hostiteľskej bunky. Štúdium z Afriky [21], [14] Taliansko, USA [ ,,,0],8] a Brazílie [29] naznačujú spojenie medzi zvýšeným počtom EPIYA C motívov a pridružených chorôb HP. Okrem toho, Sicinschi et al. [30] pozorovali pridružení medzi zvýšenými segmenty EPIYA C a prítomnosť žalúdočných prekanceróznych lézií. Na rozdiel od toho štúdie v Kolumbii [8], [31], Mexiko (J. Torres, osobné oznámenie), a Kórea [32] neboli nájdené také zoskupenia. V našej štúdii viac ako 80% všetkých vzoriek z Mexika a Venezuely boli typu ABC, a žiadny vzťah bol zjavný medzi rastom rakovina žalúdka a vyšším počtom EPIYA C motívov. Navyše nedávne štúdie [15] ukázali, že je dôležité bodových kolísanie EPIYA B motívom pre aktivity na epitelové bunky, sme pozorovali štyri non synonymické variácie v tomto motíve, ale tieto polymorfizmy nemal žiadnu súvislosť s rakovinou žalúdka. Nedávne štúdie uvádzajú dôležité pre-zápalové a pre-onkogénne aktivity ČAGA, ktoré sú nezávislé na motívy EPIYA a ktoré by mohli byť za dôležité pre ochorenie [30]; Tieto zistenia by mohlo vysvetliť nedostatok združenia C motívov s rakovinou tu hlásené aj v predchádzajúcich štúdiách. Terminál C ČAGA bielkoviny tiež obsahuje c-met motív, ktorý bol navrhnutý, aby majú niekoľko funkcií: sprostredkovať ČAGA multimerizační a membránu cielenie [33] [34], komunikovať s kinázy Par1b /Mark2 [35], a všetky tieto aktivity sú ČAGA-fosforylácie nezávislé [36]. Avšak, v našej štúdii sme nenašli významné rozdiely medzi žalúdka a karcinómu žalúdka, a to buď v sekvencii alebo v počte multimerizaci motívov. C-terminálny a N-terminálny domény ČAGA sú obaja nevyhnutné na využitie plná aktivita proteínu, aj keď majú rôzne funkcie. V poslednej dobe bolo preukázané, že N-koniec ČAGA interaguje s nádorový supresor apoptózu stimulujúce proteín p53 (ASPP2) [37]. Prítomnosť všetkých týchto interakcií medzi ČAGA a bakteriálnych a ľudských proteínov naznačujú, že to by mohlo byť veľmi ťažké pre baktérie zachovať plný rozsah biologickej aktivity v prítomnosti vysokej úrovne mutácií, z ktorých väčšina pravdepodobne viesť k strate alebo zoslabenie funkcie. Aj ČAGA Cage CAGL je špecializovaný pilus proteín, ktorý sa viaže na a aktivuje integrínu α5β1 receptor na žalúdočných epiteliálnych bunkách v prvom rade prostredníctvom svojho arginín-glycín-aspartát (RGD) motívom, vedenie správnej polohe T4SS a uľahčujúcich translokáciu ČAGA [9], [50 ]. CAGL tiež aktivuje hostiteľskou bunkou kináz fokálnej adhézne kináza (FAK) a Src, aby zabezpečili ČAGA fosforylácii v mieste vpichu, zatiaľ čo β1 integrín je vyžadované pre ČAGA vyvolané hostiteľskej bunky motility a elongácia [51]. CAGL môže byť tiež zodpovedný za HP-indukovanej hypochlórhydriou prostredníctvom aktivácie disintegrin a metaloproteázy 17 a NFkB [52]. Z dvoch CAGL V predchádzajúcej štúdii [54] sekvenčná analýza cagGamma Aj keď táto štúdia má svoje obmedzenia vo veľkosti vzorky, to je najväčší doteraz v termínoch počet cagPAI génov študovaná na hlbokú sekvenčných analýz. Niekoľko vzoriek boli stratené v dôsledku poruchy v PCR amplifikácie, ktorá môže byť v dôsledku microvariabilities z HP sekvencie.
(HP) je baktéria, ktorá kolonizuje ľudský žalúdok a môže nadviazať dlhodobú infekciu žalúdočnej sliznice. Pretrvávajúce Hp infekcie často vyvoláva zápal žalúdka a je spojená s vývojom vredovej choroby, atrofickej gastritídy, a adenokarcinóme žalúdka. Virulentný HP izoluje ukrývajúci CAG (cytotoxin spojené s génmi) patogenity Island (cagPAI), čo je 40 kb úsek DNA, ktorá kóduje zložky vylučovacieho typu IV systému (T4SS). To T4SS tvorí Pilus pre vstrekovanie faktorov virulencie do cieľových buniek hostiteľa, ako je napríklad ČAGA onkoproteínov. Analyzovali sme genetickej variability ČAGA stroje a ďalších vybraných génov HP cagPAI ( cagC
klietke
CAGL
cagT
cagV stroje a CAG Gamma
) za použitia DNA extrahuje zo zmrazených žalúdočných biopsiou alebo z klinických izolátov. Študijné predmety boli ČAGA 95 + pacientov, ktoré boli histologicky s diagnózou chronickej gastritídy alebo rakovinu žalúdka vo Venezuele a Mexiku, oblastiach s vysokou prevalenciou infekcie Hp. Sekvenačnej reakcie boli vykonávané ako Sanger a ďalšie generácie pyrosekvenování (454 Roche) metódy. Sme našli celkom 381 variantov s jednoznačným hovory pozorovali u najmenej 10% z pôvodne testovaných vzoriek a referenčných kmeňov. Porovnávali sme frekvencia týchto genetických variantov medzi rakovinou žalúdka a chronických prípadov gastritídy. Dvadsať šesť SNP (11 non-synonymá a 14 synonymické) vykazovali štatisticky významné rozdiely (P 0,05) a dva SNP, v polohe 1039 a 1041 o klietke
ukázal veľmi významnú súvislosť s rakovinou (p -hodnota = 2,07 x 10 -6), a variant Kodona bol umiestnený v VirB3 homológie domény Agrobacterium
. Výsledky tejto štúdie môžu poskytnúť predbežnú informáciu od cieľa antibiotiká k vysoko rizikových osôb, ak je účinky týchto variantov sú potvrdené ďalšie vyšetrovanie
(HP), je jedným z najčastejších chronických bakteriálnych infekcií u ľudí. Odhaduje sa, že viac ako polovica dospelej populácie na svete je nakazený týmto organizmom [1]. Medzi nimi, približne 10 až 15% infikovaných jedincov sa odhadujú na skúsenosti klinicky nepriaznivé následky, vrátane peptických vredov, adenokarcinómu žalúdka a žalúdočnej sliznice, lymfóm asociovaný s lymfoidné tkanív lymfómu (MALT) [2]. K dnešnému dňu, a to napriek rozsiahle úsilie na celom svete, čo určuje tieto variabilné klinické výsledky nebol doteraz úplne objasnený, ale veril byť kombinácia životného prostredia (napr fajčenie a stravy) [3], hostiteľskej genetika a HP faktorov virulencie [3], [4 ], [5]. Práca u nás [6] a iní podporovať, že bakteriálne faktory môžu zohrávať rozhodujúcu úlohu najviac [7], [8].
sídli v cagPAI a je zodpovedná za väčšinu z HP-spojených malígny fenotypmi: spúšťa IL-8 sekréciu priming zápalovú reakciu, podporuje bunkovú proliferáciu, rozptyl a migráciu a to buď prostredníctvom fosforylácie závislých a nezávislých mechanizmov [ ,,,0],9], [10]. CagPAI je prítomný v asi 95% z východnej Ázie izoluje a to je menej častá u izolátov z nízkorizikových západných krajín [11], [12], [13].
činnosť [14], hoci toto je sporné [15] , K dnešnému dňu, cez známych variability v N-koncovej ČAGA
génu a ďalších ostrovných gény cagPAI, došlo k veľmi obmedzené informácie o klinický význam genetických variantov mimo EPIYAs. Preto v tomto dokumente sa snažíme identifikovať varianty v génoch cagPAI cagC
( HP0546
), Cage
( HP0544
), CAGL
( HP0539
), cagV
( HP0530
), cagT
( HP0533
), a CAG Gamma
( HP0523
) gény, ktoré boli určené ako dôležité funkčné súčasti modelu bakteriálne T4SS, a je známe, že sú veľmi dôležité pre funkciu cagPAI translokácie alebo prítomný extracelulárne, čo svedčí o možnej interakcie v hostiteľských bunkách; a ČAGA,
ktorého región EPIYA bolo dôsledne koreluje s klinickým výsledkom (rakovina žalúdka) [16].
nie je dostačujúce na predvídať klinické výsledky. Okrem toho existujú náznaky, že HP odstránenie znižuje výskyt rakoviny žalúdka len u jedincov bez prekancerózne lézie. Výsledky tejto štúdie môžu poskytnúť cenné informácie zacieliť liečbu antibiotikami, aby vysoko rizikových jedincov, ak je účinky týchto variantov sú potvrdené ďalšie vyšetrovanie.
materiáloch a metódach
izoláciu DNA alebo kultúry. Expertné patológovia v neoplastické lézie v žalúdku čítať histologických preparátov.
Mexiko.
Primer konštrukcia
amplimers použité boli už skôr zverejnené [20], [21]. Všetky priméry použité pre ČAGA
cagC, klietke, CAGL, cagV, cagT stroje a CAG gama
boli najprv testované na PCR reakciách na malom počte študijných vzoriek ( n = 16) a amplifikovanej oblasti boli sekvenované s technológiou Sanger na rovnakých vzorkách pre potvrdenie špecificity amplifikácie (pozri doplnkovú tabuľku S1 pre sekvencií primerov a PCR amplifikácie podmienok).
N-terminál (630bp ), C-terminálny (poloha 2670 - 3.100) a EPIYA motívy oblasť, rovnako ako CAGL
génu boli sekvenované metódou Sanger. Sekvenačnej reakcie boli vykonané za použitia BigDyeR Terminátor Cycle Kit (Applied Biosystems, Foster City, CA, USA) za teplotných podmienok, ako sú nasledujúce: 96 ° C po dobu 2 minút, a potom 27 cyklov pri 96 ° C počas 30 s, 54 ° C počas 10 s a 60 ° C počas 4 min. Reakčné produkty sa vyzráža 2-propanolom, premyje 75% etanolom, zriedi v 25 ul vody a nanesený na ABI 3100 Genetic hranola Analyzer (Applied Biosystems). Primárne dáta sekvenovania boli analyzované pomocou programu sekvenčné analýzou (Applied Biosystems).
Bioinformatic a štatistické metódy
na 23,92% v roku cagC
, zatiaľ čo variabilita aminokyselina zaujímavo v rozmedzí od 5,69% do cagT
ukazuje najmenšej miere variácie, na 31.01% vo ČAGA
.
Cage
caggamma
a CAGL
, pričom žiadny z nich nebol umiestnený v cagC
cagT
alebo cagV
. Potom sme použili študijné ručičiek prah p = 1,31 × 10 -4 (0,05 /381) upravené pre viacnásobné porovnávania a len dva SNP, v polohe 1039 a 1041 v Cage
, vykazovali p-hodnota nižšia ako tento prah. SNP v Cage
gén (pozícia 1905) ukazuje ap hodnotu 2,55 × 10 -4 veľmi blízko študijného-múdry štatistickej významnosti.
ČAGA
polymorfizmy a typy EPIYA
SNP je uvedené v doplnkovom súboru S1). Je zaujímavé, že dvaja SNP ukazujú iný opakovanie medzi gastritídy a rakovinou prípadoch s p menšie ako 0,05, a to aj v prípade, že neboli považované za štatisticky významný vzhľadom k veľkému počtu skúšok; Jedným z nich je non-synonymný SNP (A2033G určenie aminokyselinovú zmenu T /A, pozri tabuľku 3) a jednu synonymické SNP (A2547G, pozri tabuľku 4).
PAI gény
cagT, cagV stroje a CAG Gamma
bola hodnotená 454 sekvenovanie a c AGL
podľa Sanger sekvenovania. Kompletný katalóg polymorfizmov pozorovaných v týchto siedmich génov je uvedené na doplnkovom súboru S1.
génu sme zistili 83 SNP, z ktorých 25 bolo vybraných ako je popísané vyššie. Žiadna z týchto SNP ukázala rozdielne distribúciu medzi gastritídy a rakovinových prípadov.
gén sme katalogizované 308 polymorfné miest, 97 z týchto polymorfizmy boli analyzované. C1039T a T1041G ukázala štatisticky významný iný opakovanie medzi gastritídy a rakovinových prípadov s p = 9,97 × 10 -6. Okrem toho ďalšie SNP T1905C vykazovali odlišný opakovaní s p hodnotou 2,55 x 10 -4, čo je veľmi blízko k štúdie-múdry prah. Deväť ďalších SNP vykazujú odlišný opakovanie medzi gastritídy a rakovinových prípadov s p menšie ako 0,05, šesť synonymické polymorfizmy (T1032C, C1038T, T2092C, A2097G, G2121A a A2286G) a 3 non-synonymické variantoch: A76C (aminoacidic zmena od lyzínu na glutamín), T1853C (aminoacidic zmena z valínu na alanín) a A2032G (aminoacidic prechodu z asparagínu na kyselinu asparágovú).
génu, sme pozorovali 74 polymorfizmy a 24, ktoré boli analyzované, 4 vykazovali rozdielne distribúciu medzi prípadov rakoviny a gastritídy (P menšia ako 0,05). Dvaja z nich boli non-synonymá: G166A (aminoacidic zmena alanínu na treonín) a A172G (aminoacidic zmena asparagínu na kyselinu asparágovú) a dve synonymá :. (A228G a C516T)
gén sme analyzovali 23 81 polymorfizmov pozorovaných, zatiaľ čo v cagV
génu bolo analyzovaných 11 z 61 polymorfizmy, a v oboch génov žiadna z polymorfizmu vykazovali rozdielne distribúciu medzi gastritídy a rakovinových prípadov.
génu, sme pozorovali 111 polymorfizmy, 53 z toho boli ďalej analyzované a 4 je synonymom (A195TorC, T207, C264T a A468G) a päť non-synonymné (A38G, C47G, A200 /201T, A367C a G457A) ukázala p. < 0,05 za distribúciu diferenciál medzi gastritídy a rakovinových prípadov (obrázok 1)
VirB2), ktoré tvoria hlavnú extracelulárnej štruktúru, na ktorej je hrot CAGL pripojené k interakcii s β-1 integrínu; b) základný komplexné proteíny, CagW (VirB6), CagT (VirB7), CagV (VirB8), CagX (VirB9) a opatrný (VirB10), ktoré tvoria vnútorné jadro Pilus; c) energetické faktory Cagβ (VirD4), Cagα (VirB11) klietky (VirB3 /VirB4), ATPázy dodávajúce energiu pre systém fungovať [26]. V tejto štúdii sme sekvenované cagC
( HP0546
), CAGL
( HP0539
) z Pilus, cagV
( HP0530
), cagT
( HP0533
) a CAG Gamma
( HP0523
) od hlavného komplexu, a Cage
( HP0544
) od prívodu energie enzýmy z kmeňov HP izolovaných z žalúdka a pacientov s rakovinou žalúdka. Tieto gény boli vybrané preto, že ich výrobky sú známe, že sú nevyhnutné pre funkciu T4SS a niektoré z nich sú prezentované extracelulárne H. pylori
(ČAGA, CAGL, CagC), čo naznačuje možné interakcie s hostiteľskou bunkou [27].
je najlepšie stanovená cagPAI marker virulencie, ČAGA
status sama o sebe nestačí na predpovedať klinické výsledky u vysoko rizikových skupín, kde väčšina spoločnosti HP sú ČAGA
-pozitívnych kmene. V tejto súvislosti je identifikácia nových molekulárne markery virulencie HP predpovedať riziko rakoviny žalúdka bude veľmi dôležité. Nedávny pokrok v metodike stanovenia genotypu nám umožňuje používať DNA od žalúdočných biopsiou k štúdiu microvariabilities sekvenčné HP, ktoré boli takmer výhradne študovali v kultivovaných kmeňov. Genotypizácia vyššieho počtu vzoriek žalúdočnej nám umožňuje rozšíriť cagPAI genetickú detekciu variantov s inými potenciálne významných T4SS génov. To je dôležité, pretože predchádzajúce štúdie sa zamerali predovšetkým na ČAGA stroje a užitočnosť iných cagPAI génov ako markery pre rizika ochorenia je sotva študovaný. Niekoľko štúdií sa pozrel na združenie prítomnosti cagPAI génov a chorôb, a nikto študovali polymorfizmy v génoch cagPAI, iné ako ČAGA.
je jedinečný gén, ktorý kóduje dva T4SS komponenty, VirB3 (N-koniec) a B4 (C-koniec) ako fúzny proteín [38], a B4 je najväčší ATPázy medzi niekoľkými T4SS zložiek. Generuje energiu pre proces sekrécie, a tak je nutné pre substrátu translokáciu [39] a interaguje s mnohými ďalšími T4SS proteínov, vrátane VirB2 [40]. Aj napriek svojej pomerne vnútornú lokalizáciu, hrá kľúčovú úlohu v indukcii IL-8 bola preukázaná [41], [42], [43]. Zaujímavé je, že sme pozorovali silný vzťah medzi dvomi SNP (C1039T a T1041G) na klietke stroje a rakoviny žalúdka, zistenie nie je uvedené skôr. Tieto SNP sú v pozícii jedna a tri z rovnakého kodónu a my sme vždy dodržiavať tieto dve varianty kodóny (CTT a TTG), ktoré kodifikáciu za rovnaké aminokyseliny, lyzínu. Tento variant Kodona je umiestnený v homológnej doméne s VirB3 zo Agrobacterium
. Druhý najsilnejší asociácia bola zistená v inom synonymické SNP v polohe 1905 a v tomto prípade sú dve možné kodóny boli GTT a GTC, ktoré kódujú valín. To je známa už dlhú dobu, že alternatívne synonymný kodóne sa nepoužíva s rovnakými frekvenciami a vzory využitie kodónov sa líši medzi jednotlivými druhmi [44]. využitie kodónov je vychýlený génov exprimovaných na vyššej úrovni [45], [46]. Použitie optimálnych kodónov umožňuje efektívnejšie využitie ribozómov a vedie k väčšej rýchlosti rastu [47]. Aj keď je genóm HP bolo hlásené, že neobsahujú žiadny kodóne chýb u vysoko exprimovaných génov [48], Kloster a Tang [49], ktoré možno identifikovať preferenciu vo úrovňou expresie génov, kde sa TTG kodóne prednostné cez CTT kodóne, rovnako ako VOP Kodona nad GTT. Preto na základe týchto údajov by sme mohli špekulovať, že rozdiel vo využití kodónov môžu mať vplyv na úroveň expresie génu sa silným funkčným významom pre vylučovací systém T4SS ako je napríklad Cage
.
SNP sme zistili, spojené s rakovinou žalúdka je A172G SNP (N58D) je v rovnakej polohe, v ktorej Yeh et al [53] preukázali, že súbežné prítomnosť tyrozínu v polohe aminokyseliny 58 a kyselina glutámová v polohe 59 (Y58E59) v porovnaní s kombinovanou asparágovej kyseliny (D58) a lyzínu (K59), indukuje účinnejšie korpus posun žalúdka integrínu α5β1, ktorý bol v súvislosti s žalúdočnej karcinogenéze. Sme nespozorovali tyrozínu (Y) aminokyselina v polohe 58 v každej vzorke, aj keď sme zistili, že nosiče kyseliny asparágovej (D) v tejto pozícii sú nižšie riziko karcinómu žalúdka v porovnaní s asparagínu (N) nosiča. Okrem toho sme pozorovali polymorfizmus v aminokyseliny 59 na nižšej frekvencii a bez rozdielu medzi rakovinou a gastritídy vzoriek.
génu ukázala, že skrýva typický SLT katalytická doména medzi zvyškami 33 a 165, ktorých "ES" a "AVGAY" motívy boli vysoko konzervovaný medzi ortolog enzýmov. Pozorovali sme päť nonsynonymous variantov s inou distribúciu v rakovinových a gastritídy prípadov. Tri z týchto mapy v katalytickej doméne, avšak žiaden z nich sa nachádza v najviac zachovaných častí domény.
 Apoptóza je dôležitým mediátorom patogenézy pri infekcii koronavírusom zvierat
Apoptóza je dôležitým mediátorom patogenézy pri infekcii koronavírusom zvierat
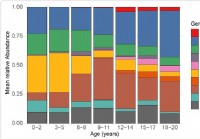 Zmena mikrobiómu horných dýchacích ciest u detí súvisiaca s citlivosťou na SARS-CoV-2
Zmena mikrobiómu horných dýchacích ciest u detí súvisiaca s citlivosťou na SARS-CoV-2
 Použitie FLUOstar Omega na štúdium nových črevných baktérií, ktoré môžu ovplyvniť naše zdravie
Použitie FLUOstar Omega na štúdium nových črevných baktérií, ktoré môžu ovplyvniť naše zdravie
 Opatrenia na zabránenie prenosu SARS-CoV-2 odpadovou vodou v chudobných oblastiach
Opatrenia na zabránenie prenosu SARS-CoV-2 odpadovou vodou v chudobných oblastiach
 Štúdia odhaľuje antivírusové účinky kurkumínu
Štúdia odhaľuje antivírusové účinky kurkumínu
 Štúdia spája konzumáciu kvasenej zeleniny s nízkou úmrtnosťou na COVID-19
Štúdia spája konzumáciu kvasenej zeleniny s nízkou úmrtnosťou na COVID-19
 Mladá krv obnovuje vitalitu u starších ľudí
Dracula Brama Stokera prežil z krvi mladých dievčat. Vedci teraz zistili, že v tejto bizarnej teórii mohlo byť niečo pravdy! Podľa genetičky z University College London Dame Linda Partridge, krv od
Mladá krv obnovuje vitalitu u starších ľudí
Dracula Brama Stokera prežil z krvi mladých dievčat. Vedci teraz zistili, že v tejto bizarnej teórii mohlo byť niečo pravdy! Podľa genetičky z University College London Dame Linda Partridge, krv od
 Črevné mikróby môžu byť spojené s depresiou
Ľudské črevo obsahuje bilióny baktérií, ktoré tvoria črevný mikrobióm alebo mikrobiotu. Nový výskum spojil zloženie tohto črevného mikrobiómu a depresívnu poruchu. Štúdia s názvom Neuroaktívny potenci
Črevné mikróby môžu byť spojené s depresiou
Ľudské črevo obsahuje bilióny baktérií, ktoré tvoria črevný mikrobióm alebo mikrobiotu. Nový výskum spojil zloženie tohto črevného mikrobiómu a depresívnu poruchu. Štúdia s názvom Neuroaktívny potenci
 Nový nástroj zaznamenáva a sleduje rast mikrobiómov
Za posledné roky, ľudský mikrobióm si získal obrovskú popularitu vďaka svojej úlohe pri formovaní vlastného zdravia. Je nevyhnutný pre rozvoj človeka, výživa, a imunitu. Preto sa mnohé štúdie zamerali
Nový nástroj zaznamenáva a sleduje rast mikrobiómov
Za posledné roky, ľudský mikrobióm si získal obrovskú popularitu vďaka svojej úlohe pri formovaní vlastného zdravia. Je nevyhnutný pre rozvoj človeka, výživa, a imunitu. Preto sa mnohé štúdie zamerali