tiivistelmä
Background
Koska markkeri Helicobacter pylori Menetelmät entropia-pohjainen laskenta havaitsemiseen käytettiin keskeisten jäämiä CagA keskeyttävät sekvenssit kuin mahasyövän biomarkkereiden. Kunkin jäännöksen, sekä kombinatorinen entropia ja tausta entropia laskettiin, ja entropian ero käytettiin kriteerinä ominaisuuden jäämiä valintaa. Ominaisuus arvot syötetään sitten Tukivektorikoneet (SVM) kanssa Radial Basis Function (RBF) ydin, ja kaksi parametrit olivat viritetty saat optimaalisen F-arvo käyttämällä ruudukkohaulla. Kaksi muuta suosittua järjestyksessä luokitus menetelmiä, BLAST- ja HMMER, käytettiin myös samat tiedot vertailtavaksi. menetelmä saavuttaa 76% ja 71% luokitustarkkuudesta Länsi- ja Itä-Aasian alatyyppejä, vastaavasti, joka suoritetaan huomattavasti parempi kuin BLAST ja HMMER. Tämä tutkimus osoittaa, että pienet vaihtelut aminohappojen niille tärkeitä jäämiä voisi johtaa virulenssin varianssia CagA kantojen seurauksena eri maha-sairauksiin. Tutkimus tarjoaa paitsi hyödyllinen väline ennustaa korrelaatio uuden CagA rasitusta ja sairauksia, mutta myös yleinen uudet puitteet havaitsemiseksi biologisista järjestyksessä biomarkkereita väestötutkimuksissa. Citation: Zhang C, Xu S, Xu D (2012) Risk Assessment mahasyövän aiheuttaman Helicobacter pylori Editor: Niyaz Ahmed, University of Hyderabad, Intia vastaanotettu 13 marraskuuta 2011; Hyväksytty: 11 huhtikuu 2012; Julkaistu: May 15, 2012 Copyright: © 2012 Zhang et al. Tämä on avoin pääsy artikkeli jaettu ehdoilla Creative Commons Nimeä lisenssi, joka sallii rajoittamattoman käytön, jakelun ja lisääntymiselle millä tahansa välineellä edellyttäen, että alkuperäinen kirjoittaja ja lähde hyvitetään. Rahoitus: Tämä työ oli osittain tuettu Yhdysvaltain National Institute of Health [lupanumeroon R21 /R33 GM078601] ja kansainvälisen vaihdon ja yhteistyön toimiston Nanjing Medical University, Kiina. Rahoittajat ollut mitään roolia tutkimuksen suunnittelu, tiedonkeruu ja analyysi, päätös julkaista tai valmistamista käsikirjoituksen. Kilpailevat edut: Kirjoittajat ovat ilmoittaneet, etteivät ole kilpailevia intressejä ole. Helicobacter pylori (H. pylori) B on gram-negatiivinen spiraali-muotoinen bakteeri elävät ihmisen mahalaukussa ja tartuttamisesta yli puolet maailman väestöstä [1], [2], [ ,,,0],3]. Viimeaikaiset tutkimukset ovat osoittaneet, että se liittyy maha- sairauksiin, kuten pohjukaissuolihaavat [4], mahahaavan [5] ja krooninen gastriitti. Vielä tärkeämpää on, se on merkittävä riskitekijä mahasyövän [6], [7], [8]. Se on luokiteltu luokan 1 ihmiselle syöpää aiheuttavaksi aineeksi Maailman terveysjärjestön vuodesta 1994 [1]. Koska markkeri H. pylori sisällä vaihtelevan alueen CagA, on olemassa joitakin erilaisia keskeyttävät sekvenssit näiden EPIYA motiiveja. Yksi kopio EPIYA plus välisekvenssin identifioidaan EPIYA segmentti. Neljä ainutlaatuista tyyppisiä EPIYA segmentit on löydetty CagA, määritellään EPIYA-A, -B, -C ja -D [11]. CagA eristetty Aasian maiden, nimetty Itä-Aasian CagA sisältää EPIYA-A, EPIYA-B ja EPIYA-D motiiveja. CagA länsimaista, EPIYA-D, on korvattu EPIYA-C. Vahvempi fosforylaatio motiivi sitova aktiivisuus EPIYA-D motiivi johtaa suurempaan morfologisia muutoksia kuin mitä EPIYA-C motiivi voi aiheuttaa infektoituneissa soluissa [11]. Juuri tämä EPIYA-D motiivi lisääntynyt sitoutumisaktiivisuutta ja tuloksena morfologiset muutokset, jotka tunnistaa sen mahdollisena tekijä selittää korkeampi esiintyvyys mahasyövän Itä-Aasian maissa [23], [24]. Aikaisemmat tutkimukset paljastivat vaihtelu määrän EPIYA aihe toistuu sekä Itä-Aasian ja Länsi CagA, joka voi vaikuttaa biologiseen toimintaan. Yamaoka et ai. [25] todettiin, että Kolumbiassa ja Yhdysvalloissa, kyky Caga-positiivisten H. pylori Koska monimutkainen ja variantti sekvenssit CagA, välisiä suhteita polymorfismi CagA ja kliinisten sairauksien tullut erittäin mielenkiintoinen tutkimus ongelma. Kuitenkin molekyylimekanismeja että taustalla eri maha- aiheuttamia sairauksia Caga-positiivisten H. pylori Tässä tutkimuksessa ehdotamme systemaattinen menetelmä analysoida paitsi määrää EPIYA kuviot CagA sekvenssit mutta myös erityisiä sekvenssikuviot puuttua alueilla. Ensimmäinen, esittelemme entropia laskenta havaitsemiseksi tähteet vaihtelevan alueen CagA kuten mahasyövän biomarkkereita. Sitten käytämme ohjattu oppiminen menettely luokitella syöpää ja ei-syöpä käyttämällä tietoja havaittujen jäämien CagA kuin ominaisuuksia. Valitsemme tukivektorikoneet (SVM) binääriseksi luokittelija ja vertaamme menetelmä muiden kanssa. Lähestymistapamme ei vain todistaa hypoteesia, että sekvenssi vaihtelevan alueen CagA sisältää tietoa erottaa eri sairauksien, mutta myös hyödyllinen väline ennustaa korrelaatio romaani CagA kantoja ja sairauksia ja havaita biomarkkereiden samoin. Methods Data Esikäsittely Perustuen edelliseen kuvaus Ref. [15], me nimetty EPIYA motiivi ja seuraavan välissä alueilla R1, R2, R3, R3 ', R4 ja R4' (kuvio 1). Kuvio 2 esittää kannan välinen suhde EPIYA motiivi (R1) ja muiden välissä alueilla käyttäen CagA tyyppiä A-B-D (Aasian alatyyppi) ja A-B-C (Western alatyyppi) esimerkkeinä. R2 on suhteellisen säilynyt poikki molemmat alatyyppiä, mutta on merkittäviä eroja välissä alueiden R3 ja R3 ', välillä sekä R4 ja R4'. Itä-Aasian alatyyppi ja Länsi alatyyppi käsiteltiin kahdesta riippumattomasta. Heidän data käsiteltiin sitten ja tulokset analysoitiin kussakin ryhmässä erikseen. Kaikki välissä alueet poimittiin CagA sekvenssit ja panna vastaaviin alatyyppi ryhmiä, ja sitten usean sekvenssin rinnastukset haettiin kustakin ryhmästä erikseen käyttämällä Clustal X versio 2.0.3 [29]. Sekvenssit profiilit (kuva 1) rakennettiin käyttäen Weblogo 3 [30]. Koska CagA liittyy lähes kaikkiin maha- sairauksia ja yksinkertainen analyysi EPIYA motiivin toistojen ei tuottanut tilastollisesti merkittäviä eroja näiden tautien tieto siitä tietyn taudin saattaa olla piilotettu muuna alueilla. Tämä tutkimus edellyttää, että on olemassa joukko jäännöksiä tai jäämiä yhdistelmiä, jotka voivat olla hyödyllisiä markkerina tietyn taudin. Tämä tutkimus keskittyy mahasyövän ja käyttää syövän /ei-syöpä ryhmät esimerkkipolkuna. Perustuu linjassa sekvenssit kunkin välissä alueen erityiset tähteet tunnistettiin vertaamalla ero kombinatorisista entropian [31] välillä syöpä ja ei-syöpä ryhmiä. Tämä menettely sisältää seuraavat vaiheet: Ensinnäkin jaamme annettu monirinnastukset kaikkien välissä olevien alueiden kahteen ryhmään: mahasyövässä ryhmä ja ei-syöpä ryhmä. Kunkin sarakkeen monirinnastukset laskemme taustan entropia (Eq. 1) ja kombinatorinen entropia (Eq. 2), kuvataan seuraavasti: (1) jossa edustaa lukumäärää sekvenssien ryhmä k Sitten entropia ero kombinatorisista entropia ja taustan entropia on laskettu: (3) B- Kuvio 3 esittää entropian käsite kolmella ääritapauksissa. Tapauksessa P1, aminohapot "satunnaisesti ja tasaisesti jakautunut" kaikkien ryhmien ja ei ole merkittävästi konservoitunut malli tässä asennossa. Asia P2 edustaa "maailmanlaajuisesti konservoitunut" malli ja kaikki aminohapot ovat samat kaikkialla molemmissa ryhmissä. Tapauksessa P3, tiettyjä aminohappoja on vain konservoituneita erityisesti ryhmissä, ja eri ryhmillä on erilaiset aminohappoja. Kutsumme tätä asia "paikallisesti säilynyt". mukaan laskennan tulokset entropian ero edellä kolmessa tapauksessa kombinatorisista entropia on niin "maailmanlaajuisesti säilyneitä" ja "paikallisesti säilyneitä tapaukset. For "satunnaisesti ja tasaisesti jakautunut" tapauksessa saa maksimiarvon. Voimme erottaa "säilytetty" ja "satunnaisesti ja tasaisesti jakautunut tapaukset perustuu kombinatorisista entropia, mutta se ei auta poimia" paikallisesti konservoitunut "tapauksessa kaikista" säilynyt "tapauksissa. Kun otamme huomioon taustalla entropia samanaikaisesti, saa maksimiarvo, 0 ja keskisuurten arvo "satunnaisesti ja tasaisesti jakautunut" tapauksessa "maailmanlaajuisesti säilytetty" tapauksessa "paikallisesti säilynyt" tapauksessa vastaavasti. Lopuksi erot edellä mainituista kolmesta tapaukset ovat :,, ja saa minimiarvon. Siten entropia ero on asianmukaisen mittauksen havaitsemiseksi "paikallisesti konservoitunut" järjestyksessä malli. Edellä esitetyn perusteella laskelma, se voidaan määrittää, että oikea ryhmittymä voi minimoida entropian ero näiden jäämien jotka kuuluvat "paikallisesti säilytetty" tapaus. Suoritettava testi, yksi sekvenssi valitaan ja loput sekvenssit on jaettu mahasyövän ryhmä ja ei-syöpä ryhmä. Kaikkien valittujen tähteiden, valittu sekvenssi laitetaan mahasyövän ryhmä laskea entropian erotus, ja sitten se on sijoitettu ei-syöpä ryhmä saada vastaava entropian erotus. Lopuksi saadaan kaikkien valittujen tähteitä, joita käytetään ominaisuus entropia. Dataset. Haimme National Center for Biotechnology Information (NCBI ), Swiss-prot /Tremble ja DDBJ proteiini tietokantaan ja saadaan 535 kantoja H. pylori Kuva 4 esittää työnkulun luokittelun /ennuste menettely: Bootstrapping. merkittävä kysymys rakentamisessa luokitusmalli tässä tapauksessa on suuri ero näytteen koot välillä syöpä ja ei-syöpä ryhmiä, jotka voivat aiheuttaa bias luokittelutuloksissa. Bootstrapping menettelyä sovellettiin tämän asian. Kussakin alatyyppi ryhmässä, jokaista koulutukseen /testi aineistoja, kaikki ei-syöpä näytteet mukana, ja sitten kantoja jatkuvasti vedetään syöpään ryhmästä satunnaisesti saapumiseen asti samankokoinen kuin syöpä ryhmä. Tässä tapauksessa kaikki saatavissa tietoja käytettiin vaikka syöpänäytteissä hyödynnettiin useita kertoja koska niiden pienempi koko verrattuna ei-syöpä ryhmä. Tätä menettelyä sovellettiin viisi kertaa tuottaa viisi itsenäistä koulutusta sarjaa kunkin testijakson. Luokittelu /ennuste tulos on keskiarvo Näiden viiden itsenäisen tuloksia. Koska datan koko on pieni, jätettävää one-out (LOO) rajat validointimenettelyllä esitettiin. Tämä ei ole vain arvio lajittelijan suorituskykyä koulutus /testituloksia, mutta myös arvio ennusteen valtaa uusia tapauksia. Valitsimme SVM kuin binary luokittelija ja käytti ominaisuus-entropia vektorit kouluttaa ja testata luokittelija. Jos kyseessä on kahden luokan pehmeä marginaali luokitusta, päätösfunktion on painotettu lineaarinen yhdistelmä määritellään seuraavasti: (4) missä edustaa käyttäjän määrittämä ytimen toiminto, joka mittaa yhtäläisyyksiä tulon piirrevektorin ja piirrevektorit koulutuksessa aineisto. on osoitettu paino koulutusta piirrevektorin ja osoittaa, onko CagA kanta on merkitty positiivinen luokka (+1) tai negatiivinen luokka (-1). Primal optimointi ongelma on muodoltaan: minimoida (5) edellyttää (6) missä. m on kokonaismäärä kantoja. on löysä muuttuja, joka mittaa aste Luokitteluvirheillä peruspisteen. on kustannus parametri, joka mahdollistaa kaupankäynnin pois koulutukseen virhe vastaan mallin monimutkaisuutta. w on normaali vektori ja b on offset. Kun verrataan tuloksia polynomin, tanh ja Gauss radial perusteella ytimet saatu tulos kanssa RBF ytimen toimi parhaiten, jos Gaussin Radial Basis ytimet (RBF :) ovat yleiskäyttöisiä oppiminen jos ei ole etukäteen tietoa data. SVM Light paketti (http://svmlight.joachims.org/) [32] käytettiin rakentaa sovelluksen. Parametrit ja oli viritetty saada paras malli harjoitustietosivut kuten esitetään seuraavassa. Kaikki muut SVM parametrit asetetaan oletusarvoihinsa. Jotta voitaisiin arvioida suorituskykyä luokittelija, erilaisia suorituskykyä toimenpiteet toteutetaan: tarkkuus, herkkyys ja spesifisyys. Todellinen positiivinen (TP) on syöpään liittyvien sekvenssin pitää sellaisena, kun taas väärä positiivinen (FP) on ei-syöpään liittyvät sekvenssin luokiteltu syöpään liittyvien, väärä negatiivinen (FN) on syöpään liittyvät sekvenssin luokitella ei -cancer liittyvät ja todellinen negatiivinen (TN) on ei-syöpään liittyvät sekvenssin luokiteltu ei-syöpään liittyvät. Tarkkuus, herkkyys (Sn), spesifisyys (Sp) ja Matthews korrelaatiokerroin (MCC) luokituksen määritellään seuraavasti: (7) (8) (9) (10) Koska on olemassa vain kaksi parametrit RBF ytimen ja ne ovat riippumattomia, haimme grid-haku määrittää optimaaliset parametrit luokittelija. Käytimme harmoninen keskiarvo herkkyyden ja spesifisyyden kuin kohdefunktion optimoida mallin koulutusta asetettu, joka määritellään seuraavasti: Tulokset tapahtuvan jäämien ja ominaisuus laskeminen taulukossa 1 luetellaan kaikki havaitut keskeiset tähteet laskemalla entropian ero jokaisessa välissä alueen sekä Länsi-ja Itä-Aasian alatyyppejä. Tosin jotkut maantieteellisiä muunnelmia CagA sekvenssit läntisen ja Itä-Aasian alatyyppejä, joitakin yhteisiä jäämiä saattaa vielä löytyä erottaa syövän ja ei-syöpä ryhmiä. Se ehdottaa, että nämä jäämät voivat olla hyvin tärkeitä määritettäessä virulenssin CagA ja suhde CagA ja tiettyjä sairauksia. Jäännös asemat on esitetty kuvassa 5. Edellisessä tutkimuksessa [27] osoittaa, että eri EPIYA segmentit voivat sitoutua eri kinaasien, esim EPIYA-R2 ja EPIYA-R3 /R3 'sitoutuvat C-terminaalin Src (Csk), kun taas EPIYA-R4 ja EPIYA-R4', sitoutuvat SHP-2 kinaasin aiheuttaa hummingbird-fenotyyppi. CagA-Csk vuorovaikutus down-regulation CagA-SHP-2 signalointi joka häiritsee solun toimintoja, jotka ohjaavat virulenssiin CagA. On havaittu, että useimmat havaitut jäämät kuuluvat R2 ja R3 /R3 'alueet ja muutaman tähteen R4 /R4 alueet on havaittu. Tämä voi johtua siitä, että R4 /R4 'on enemmän konservoituneita sekvenssin kuin R2, ja R4 /R 4' on lyhyempi kuin R3 /R3 '. Ehdotamme, että eri jäännös kuvioita R2 tai R3 /R3 'alueet saattavat muuttua kykyä alas säätelevä CagA-SHP-2 signalointi ja siksi muuttaa virulenssiin CagA. Ren et ai. havaitsi, että CagA multimerizes nisäkässoluissa [33]. Tämä multimerisaation on riippumaton sen tyrosiinifosforylaatiota, mutta se liittyy "FPLxRxxxVxDLSKVG" motiivi, joka on nimetty CM motiivi R3 'välissä alueella. Koska Multimerisaatiodomeeni on edellytys CagA-SHP-2 signalointikompleksiin ja myöhempi vapautuminen SHP-2, CM motiivi on tärkeä rooli Caga-positiivisten H. pylori Vaikka avaimen ainejäämää voi paljastaa joitakin eroja syövän ja ei-syöpä ryhmät, mikään yksittäinen tähde voi olla markkeri syövän, kuten on esitetty kuviossa 5. Tämä tutkimus arvioi, että yksi erityinen yhdistelmä kaikista tai osittain havaittu jäämiä saattaa olla suuri korrelaatio tietyn sairauden. Voit tarkistaa useita lineaarisia tilastollisia malleja, esim. lineaarinen regressio ja logistinen regressio, levitettiin havaitut ominaisuudet voidaan arvioida, että on tärkeää kunkin jäännöksen ja korrelaatio valittujen tähteiden ja syöpä. Kuitenkaan mikään edellä mainituista malleista pystyivät tuottamaan tulos on tilastollisesti merkitsevä. Koska ominaisuuksia ei voida asentaa yksinkertaisella lineaarisia malleja ennustamaan syöpä, soveltamalla koneoppimisen menetelmä analysoida ja luokitella nämä tiedot ovat tarpeen. Käyttämällä Länsi alatyyppi ryhmä esimerkissä löysä grid-haku ensin suoritettiin ja (kuvio 6A) ja totesi, että paras on noin saada korkein F arvoa LOO ristivalidointi korko 76%. Sitten hienompi ruudukkohaulla tehtiin naapuruston ja parempaa F-arvo saavutettiin 79,7% LOO ristivalidointi osoitteessa. Samaa menettelyä käytetään Itä-Aasian alatyyppi ryhmä ja paras LOO ristivalidointi korko 72,6% saavutettiin. Koska ei ole aikaisempia tutkimuksia tai laskennallisia menetelmiä samasta aiheesta, tehokkuuden arviointiin tämän tutkimuksen uusi menetelmä on vaikeaa. Arvioimaan tietosisältöä sekvenssien suhteen heidän vaativille valta ennustaa syövän, satunnainen sekoitus meneteltiin rakentaa kontrolliryhmään. Ensinnäkin, kaikki sekvenssit Länsi alatyypin asetettiin yhdessä rakentaa naissekvenssipoolin. Toiseksi, meidän sattumanvaraisesti sama määrä sekvenssejä, kuten syövän ryhmä sekvenssi allas ja käsiteltiin muiden sekvenssien kuin ei-syöpä ryhmä. Sitten koko koulutuksen menettelyä sovellettiin vasta sekoitetaan datan löydettäisiin paras. Edellä mainitut toimenpiteet toistettiin viisi kertaa tuottaa viisi itsenäistä sekoitetaan aineistoja. Yksi korkein F On pääasiassa kolmeen ryhmään järjestyksessä luokitus menetelmät: ominaisuus perustuu järjestyksessä etäisyys perustuva ja malli perustuu. Menetelmä, että me tässä asiakirjassa kuvatuin tavoin kuuluu ominaisuus-pohjainen luokkaan. Valitsimme kaksi suosituinta järjestyksessä luokittelu työkaluja edustajana menetelmiä muiden kahteen ryhmään vertailuun. BLAST [36] valittiin sekvenssin etäisyyden perusteella luokkaan, koska se on laajimmin käytetty sekvenssin vertailun avulla. Sillä mallipohjaisia tyylinen HMM malli on tyypillinen menetelmä sekvenssianalyysillä ja sen laajalti käytetty työkalu, HMMER [37], valittiin. Luokittelusta menettely sekä BLAST ja HMMER käytimme oletusparametrit työvälineistä, sovelsi samaa LOO ristivalidointi kuin meidän menetelmää, ja käytetään samaa arviointia kaavat lueteltu menetelmät -kappaleessa. Taulukko 2 luetellaan luokittelun tulokset kaikilla tavoilla. SVM menetelmä toimii huomattavasti paremmin kuin kaksi muuta mallia. BLAST saavuttaa lähellä tarkkuus on Entropia-SVM menetelmä, mutta se ennusti monia vääriä negatiivisia, joissa alhainen herkkyys. HAMMER saavuttaa suuri herkkyys, mutta vain vähän spesifisyys. Ottaen F luokitustulos ja ääriviivaa (kuva 6) voimakkaasti tukemaan hypoteesi, eli tiedot valitun tähteiden välissä alueilla voidaan luokitella suhde CagA sekvenssien ja mahasyövän, vaikka ero profiilien syövän ja ei-syöpä ryhmiä ei ole kovin vahva. vertailu eri sairaudet H. pylori Koska useimmat tiedot kerättiin julkisista tietokannoista ilman tarkkaa diagnoosia tiedot, ennen kuin meidän tapa CagA datan, me käsin kuratoi tauti merkinnät kaikkien kantojen tarkistamalla kirjallisuudessa. Taulukko S1 luetellaan jakaumia merkittävien sairauksien sekä Länsi- ja Itä Asain alatyypin ryhmiä. Rajoituksista johtuen kannan määrän joidenkin sairauksien, kuten atrofinen gastriitti (AG) ja mahahaava (GU), me lopulta poimitaan krooninen gastriitti (CG) ja pohjukaissuolihaava (DU) kuin verrokkiryhmässä arvioitavaksi. DU ryhmä Itä-Aasian alatyypin sisältää 79 kantoja, ja bootstrapping menettelyä sovellettiin kaikkiin muihin ryhmiin, jotta sama määrä kantoja kuten Itä-Aasian DU ryhmä. Tämä vaihe takaa kaikille vertailuissa samassa mittakaavassa, koska arvo kombinatorisista entropia riippuu sekvenssien määrä. Käytimme kaava (3) voidaan laskea entropian ero kunkin aseman välillä GC ja CG /DU ryhmiä, ja lisättiin sitten kaikki entropia eroja koko erotus GC ja CG /DU ryhmiä, kuten on esitetty taulukossa S2. Vertaamalla tuloksia kahden ryhmään samassa maantieteellisellä alatyypin (Itä-Aasian tai Western alatyyppi), se on sopusoinnussa kliinisen että gastriitti on vahvempi suhteet syöpään kuin DU [39] (yleensä, gastriitti tapauksissa saattaa sisältää joitakin ilmoittamatta tai diagnosoimatta krooninen atrofinen gastriitti ja suoliston metaplasiaa tapauksissa, joihin potilaat on suuri riski kehittää GC). Tarkastelemalla saman taudista parin kahden maantieteellisen alatyyppiä, se selitti myös virulenttia ero Itä-Aasian ja Länsi-alatyyppejä. Lisäksi koska suuri samankaltaisuus eri tautiryhmistä Itä-Aasian alatyyppi, jopa enemmän tietoa, emme voi vielä saavuttaa sama luokitustarkkuudesta kuin Länsi-alatyyppiä ryhmä. Edellä olevien tulosten CagA sekvenssit osoittavat potentiaalia erottaa useita maha-sairauksia. Jotta voitaisiin arvioida luokituksen Käytimme DU ryhmä korvata ei-Cancer ryhmä, ja sitten soveltaa koko luokittelu menettelyn uudelleen ilman bootstrap, koska nämä kaksi tautia ryhmää on vastaavia kokoja. Taulukossa S3 esittää luokittelun tuloksia. Vaikka kliinisestä näkökulmasta, DU on negtive korrelaatio GC kaikkien maha- sairaudet [40], luokittelu suorituskykyä kaksi alatyyppiä ryhmää oli vain hieman. Näin syöpään liittyvien CagA kannat saattaa olla joitakin ainutlaatuisia sekvenssikuviot verrataan kaikkiin muihin maha-sairauksia. Näin ollen, tuning osajoukko kontrolliryhmä ei ehkä pysty parantamaan luokittelun tarkkuus. Vaikka tutkimukset osoittavat, että on olemassa sekvenssi markkereita erottaa syövän ryhmä ja ei-syöpä ryhmä , suuret profiilit näiden kahden ryhmät ovat liian samanlaiset erottaa käyttäen perinteisiä menetelmiä, koska CagA sekvenssit yleisesti erittäin konservoituneita. Siksi olemme keskittyneet tunnistaa informatiivinen jäämät, tiedon kvantifiointiin näiden valittujen tähteiden, ja sitten käyttää sitä suunnitella luokittelija, joka voi ennustaa, onko uusi sekvenssi kuuluu syöpä ryhmä tai ei-syöpä ryhmä. Tämä menetelmä ei vain valottaa suhteista CagA sekvenssit ja mahasyövän, mutta voi myös tarjota hyödyllinen väline mahasyövän diagnosoimiseksi tai prognoosi. mekanismit H. pylori
, Sytotoksiini liittyvä geeni A (Caga) on ilmoitettu olevan merkittävä virulenssia aiheuttava tekijä maha- sairauksia. Kuitenkin molekyyli mekanismeihin, jotka ovat kehitetään erilaisia maha- aiheuttamien sairauksien Caga-positiivisten H. pylori
infektio ei vielä tunneta. Nykyiset tutkimukset rajoittuvat arviointiin korrelaatio sairauksien ja lukumäärän Glu-Pro-Ile-Tyr-Ala (EPIYA) motiiveja CagA rasitusta. Edelleen ymmärtää suhdetta CagA sekvenssin ja sen virulenssi syöpään, ehdotimme järjestelmällistä entropian perustuva lähestymistapa tunnistaa syöpään liittyvien tähteiden muuna alueilla CagA ja työssä valvottu koneoppimisen menetelmä syövän ja ei-syöpätapausta luokitus.
Johtopäätös
käyttäminen CagA Sequence Merkit. PLoS ONE 7 (5): e36844. doi: 10,1371 /journal.pone.0036844
Johdanto
The Sytotoksiini liittyvän geenin A (Caga) on ilmoitettu lisäanalyysillä olevan merkittävä virulenssitekijäksi. H. pylori
jotka kantavat Caga geeni lisäävät riskitekijä maha sairauksien kolme taittuu yli Caga-negatiivisia kantoja [6], [9], [10]. CagA, joka koodaa CAGA-geeni, on 125-140 kDa: n proteiini. Se sisältää 1142-1320 aminohappoja ja on vaihtelevan alueen C-pään alueen, jonka eri lyhyitä sekvenssejä (kuten EPIYA motiivi) toista 1-7 kertaa. Sen jälkeen H. pylori
asettumaan pinnalla mahalaukun epiteelin, CagA voidaan translokaatio osaksi mahan epiteelisolujen kautta tyypin IV eritystä järjestelmään. Kun ruiskutetaan isäntäsoluun, CagA paikantuu plasmamembraanin ja ne voidaan fosforyloituu Src-perheen tyrosiini- kinaasien erityisistä tyrosiinitähteissä viiden aminohapon (EPIYA) motiivi [11], [12], [13] , [14]. Tyrosiinifosforyloitunut CagA sitten sitoutuu spesifisesti SHP-2 tyrosiinifosfataasin 11,15 aktivoimaan rylaasia, joka aiheuttaa kasautumisilmiöön joka häiritsee signaalinvälitysreitin isäntäsolun, joka johtaa uudelleenjärjestelyn isäntäsolun solun tukirangan ja muodostumista kolibri fenotyyppi [11], [16]. Samalla kautta aktivoimalla mitogeeniaktivoidut proteiinikinaasi (MAPK), solunulkoinen signaali säädelty kinaasi (ERK) [17] ja polttovälin tarttuvuus kinaasi (FAK), CagA voi myös aiheuttaa soluhajotusprosessin ja infiltratiivinen kasvaimen kasvua [18], [19 ], [20], [21]. Tällainen prosessi tekee CagA tärkein virulenssin tekijä H. pylori
[22].
aiheuttamaan mahalaukun limakalvon surkastumista ja suoliston metaplasiaa voi liittyä määrän EPIYA motiiveja CagA rasitusta. Hopea et ai. [16] tuli samaan johtopäätökseen myöhemmin. Vastoin lausuntoja julkaissut Lain ym. [26], joka perustuu havaintoihin mitään suhdetta lukumäärän EPIYA motiiveja CagA rasitusta ja kliinisen taudin kuluessa 58 isolaatteja Taiwan. Ottaen huomioon koko ja maantieteellisiä rajoituksia näiden tutkimusten onko tämä johtopäätös on kyseenalainen. Sen lisäksi, että numero EPIYA motiivin toistoja, sekvenssi ero kantojen vaihtelevien alueiden voi myös aiheuttaa merkittäviä eroja virulenssin, jotka voivat koskea eri patogeenisiä kyvyt H. pylori
[27].
infektio ei vielä tunneta. Tähän asti useimmat tutkimukset ovat vielä rajoitettu löytö tai arviointiin korrelaatio määrä CagA EPIYA motiiveja ja sairaudet [28].
tapahtuvan jäämien
. ilmaisee tähteiden lukumäärä tyyppiä sarakkeessa i
ryhmän k
. on tähteiden lukumäärä tyyppiä sarakkeessa i
. edustaa kokonaismäärä sekvenssien linjaus. (2) missä.
Feature-entropia laskeminen
luokittelu CagA Jaksot
CagA proteiinia. Joukossa on 287 Itä-Aasian alatyypin kantoja ja 248 Länsi alatyypin kantoja. Itä-Aasian alatyypin ryhmä, 47 pois 287 kannat ovat mahalaukun syöpäpotilaita ja loput muista sairauksista. Länsi-alatyyppiä ryhmä, meillä on 37 kantoja mahan syöpäpotilaita, ja jäännökset ovat muista sairauksista tai tavanomaista valvontaa, mukaan lukien 24 kannat vapaaehtoisilta, joiden terveys (sairaus) tila oli tuntematon.
työnkulku.
Ristiinvalidointi.
SVM.
toiminnan arviointi.
(11)
-välitteisen mahalaukun synnyssä. Kun useita CM motiiveja H. pylori
kannat paljon todennäköisesti liittyy vakavia maha- sairauksiin [33], [34], mutta tämä havainto voi selittää, miksi eri maha- sairauksia voidaan kehittää tarkka sama määrä CM motiiveja. Tutkimuksemme havaittu kaksi jäämien CM motiivi R3 'välissä alueella, joka saattaa johtaa muutokseen multimerointia, mikä muuttaa virulenssiin CagA. Tämä on sopusoinnussa aiemman löytö [35], että sekvenssi ero Aasian CM ja Länsi CM määrittää sitoutumisaffiniteetin välillä CagA ja SHP-2.
Parameter Training Luokitusperusta
arvo, mikä vastaa 46,6% valittiin ja sen Ääriviivakaavioissa on esitetty kuviossa 6B. Tämä satunnaisesti laahustavat arvioinnissa sovellettiin myös Itä-Aasian alatyyppi tietojen ja parhaiden F
arvo oli 54,3%. Verrattaessa kahden tontin osoittaa merkittävää eroa F
arvojen datan oikean ryhmittely syöpä ja ei-syöpätapausta koulutukseen ja paras arvotaan uudestaan dataa. Tulos viittaa siihen, että välissä alueet ovat informatiivisia erottamaan syövän ja ei-syöpä ryhmät ja meidän menetelmä voi käyttää tietoja tehokkaasti.
Classification suorituskyky
arvot ja MCC
arvojen tulosennusteita BLAST ja Hammer ovat lähes satunnaisia.
infektio liittyy useimmat maha- sairauksiin, joista mahasyöpä on ankarin aiheuttavat yli 700000 kuolemantapausta vuosittain maailmanlaajuisesti [38]. Koska H. pylori
on tärkein riskitekijä mahalaukun syövän (GC), löytö mekanismi H. pylori
välittävä GC tulee etusijalla tehtävä tällä alalla. Verrattaessa muihin sairauksiin, diagnoosi tiedot GC julkisista data on suhteellisen tarkka, ja se on toinen tärkeä syy keskittyä GC tässä asiakirjassa. Tutkimuksemme eivät rajoitu GC, vaikka. Olemme myös yrittäneet arvioida suhteita varianssi CagA sekvenssit ja eri sairauksien.
Keskustelu
aiheuttaen eri maha- taudit ovat vielä epäselviä, mutta on todennäköistä, että eri maha- aiheuttamia sairauksia H. pylori
infektio jakaa joitakin sekvenssikuviot muuna alueilla. Pienet vaihtelut aminohappojen näissä tärkeitä jäämiä voisi johtaa virulenssin varianssia CagA kantojen seurauksena eri maha-sairauksiin. Vaikka CagA voisi olla markkeri paljastaa mahdolliset syöpäriskin käyttäen CagA yksin erottaa kaikki maha-sairauksia ei ole realistinen. Tulevaisuuden tutkimus, tulemme kehittämään uusia malleja, jotka erottavat eri maha- tauteja Caga ja muita geenejä.
tukeminen Information
Taulukko S1.
lukumäärä kantoja kussakin tauti.
doi: 10,1371 /journal.pone.0036844.s001
(DOC) B Taulukko S2.
Yhteensä entropia ero mahasyövän ja kaksi muiden sairauksien ryhmää.
doi: 10,1371 /journal.pone.0036844.s002
(DOC) B Taulukko S3.
Luokittelu suorituskyvyn välillä mahasyövän ja pohjukaissuolihaava ryhmiä sekä Länsi- ja Itä-Aasian alatyyppejä.
doi: 10,1371 /journal.pone.0036844.s003
(DOC) B
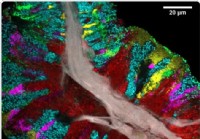 Yksityiskohtainen kartta ihmisen kielen mikrobiomista
Yksityiskohtainen kartta ihmisen kielen mikrobiomista
 Punaisen lihan vaihtaminen kasviperäisiin lihavaihtoehtoihin vähentää sydän- ja verisuonitautien riskiä
Punaisen lihan vaihtaminen kasviperäisiin lihavaihtoehtoihin vähentää sydän- ja verisuonitautien riskiä
 Tutkijat manipuloivat suoliston bakteerilajeja ruokavalion avulla
Tutkijat manipuloivat suoliston bakteerilajeja ruokavalion avulla
 Suuhygienia ja COVID-19:n vakavuus-yhteys
Suuhygienia ja COVID-19:n vakavuus-yhteys
 Tutkijat löytävät uuden tavan suojautua sairauksilta MS -mallissa
Tutkijat löytävät uuden tavan suojautua sairauksilta MS -mallissa
 Tutkijat käyttävät faagiterapiaa menestyksekkäästi alkoholisairauden hoitoon
Tutkijat käyttävät faagiterapiaa menestyksekkäästi alkoholisairauden hoitoon
 Mikrobit voivat ennustaa kuolemaan johtavia seurauksia hengitetyissä COVID-19-potilaissa
Läsnäolo Mycoplasma salivarium hengitettynä COVID-19-infektion sairastavien potilaiden alemmissa hengitysteissä liittyy lisääntynyt todennäköisyys kuolla. Tulos oli osa molekyylitutkimusta, jossa ta
Mikrobit voivat ennustaa kuolemaan johtavia seurauksia hengitetyissä COVID-19-potilaissa
Läsnäolo Mycoplasma salivarium hengitettynä COVID-19-infektion sairastavien potilaiden alemmissa hengitysteissä liittyy lisääntynyt todennäköisyys kuolla. Tulos oli osa molekyylitutkimusta, jossa ta
 Tutkijat kehittävät lähestymistavan suolen tulehdusta vastaan rokottamiseen
Tulehduksellinen suolistosairaus (IBD) on kattava termi, joka kuvaa monia sairauksia, joihin liittyy krooninen ruoansulatuskanavan tulehdus, mukaan lukien haavainen paksusuolitulehdus ja Crohnin tauti
Tutkijat kehittävät lähestymistavan suolen tulehdusta vastaan rokottamiseen
Tulehduksellinen suolistosairaus (IBD) on kattava termi, joka kuvaa monia sairauksia, joihin liittyy krooninen ruoansulatuskanavan tulehdus, mukaan lukien haavainen paksusuolitulehdus ja Crohnin tauti
 E. coli -bakteerin leviäminen huonon wc -hygienian vuoksi,
ei ruoan kautta Uusi tutkimus julkaistiin vuonna Lancetin tartuntataudit lokakuuta 22, 2019, sanoo, että yksi yleinen superbug, joka aiheuttaa yli 5, Englannissa vuosittain 000 ruokamyrkytystapausta
E. coli -bakteerin leviäminen huonon wc -hygienian vuoksi,
ei ruoan kautta Uusi tutkimus julkaistiin vuonna Lancetin tartuntataudit lokakuuta 22, 2019, sanoo, että yksi yleinen superbug, joka aiheuttaa yli 5, Englannissa vuosittain 000 ruokamyrkytystapausta