Maintenant, un groupe de scientifiques a mis au point une nouvelle méthode pour une analyse rapide et à grande échelle de chaque type de bactérie dans l'intestin, ainsi qu'une liste des espèces trouvées dans l'intestin humain sain par type et nombre (GutFeelingKB), et un nouveau modèle de rapport appelé Fecal Biome Population Report (FecalBiome) qui facilitera la compréhension exacte de ce qui se passe dans l'intestin.
 Structure cristalline de la bêta-galactosidase putative de Bacteroides fragilis. Crédit d'image:Instituts nationaux de la santé
Structure cristalline de la bêta-galactosidase putative de Bacteroides fragilis. Crédit d'image:Instituts nationaux de la santé Micro-organismes, ou microbes, existent depuis très longtemps, et façonner à la fois l'environnement externe et interne des êtres humains. Le mot « microbiome » a été inventé par Joshua Lederberg en 2001, comme moyen d'attirer l'attention des scientifiques sur les multiples interactions entre les microbes qui habitent sur et dans notre corps, et leur interaction avec notre physiologie humaine. Le terme est désormais défini comme « une communauté multispécifique de micro-organismes dans n'importe quel environnement :hôte, habitat, ou écosystème. Loin d'être de simples envahisseurs acharnés à notre destruction, le microbiome humain comprend tout un monde vivant à lui seul, apportant un ensemble complet et très diversifié de gènes qui interagissent et changent, et ont également un impact sur la santé humaine. Ce mélange génétique microbien est appelé le métagénome. Le Human Microbiome Project (HMP) a décollé en 2008 et a contribué à catalyser une meilleure caractérisation et une meilleure compréhension du fonctionnement de ces communautés.
La recherche sur le microbiome intestinal nécessite une collecte et une analyse de données précises à haut débit, ainsi que des installations pour intégrer les données traitées de manière organisée pour le stockage, partage et accès entre les groupes de recherche. La plupart des études antérieures se sont concentrées sur des gènes ou des groupes d'organismes spécifiques, laissant de côté de larges segments des génomes microbiens. Aussi, différentes normes de référence ont conduit à une variété d'énoncés sur la composition de l'intestin.
 Bactéries Bacteroides fragilis, l'un des composants majeurs du microbiome normal de l'intestin humain, Illustration 3D Crédit :Kateryna Kon / Shutterstock
Bactéries Bacteroides fragilis, l'un des composants majeurs du microbiome normal de l'intestin humain, Illustration 3D Crédit :Kateryna Kon / Shutterstock En réalité, la plupart des études sur le métagénome n'utilisent qu'un petit ensemble de référence de séquences d'acides nucléiques provenant de microbes ou de gènes microbiens déjà sélectionnés. Ceci est dû à la difficulté de coupler les données expérimentales avec la base de données complète de nucléotides disponible avec le NCBI (NCBI-nt). Cependant, de nouveaux algorithmes peuvent désormais utiliser ces derniers pour permettre une analyse plus précise des données expérimentales afin d'obtenir un profil d'abondance microbienne.
Le présent travail s'appuie sur cette base pour former une base de connaissances Gut Feeling - GutFeelingKB - avec des échantillons d'un ensemble sain de participants. Ces échantillons de microbiote intestinal ont été séquencés pour obtenir une image de ce à quoi ressemble un métagénome intestinal sain. Le numéro d'échantillon a été rempli à l'aide de 50 séquences supplémentaires sélectionnées au hasard dans le HMP.
Les chercheurs ont également collecté des séquences contiguës assemblées, ou contigs, qui ne correspondent à aucune séquence NCBI-nt mais peuvent être détectés dans des échantillons fécaux sains. Les contigs sont donc de la matière noire, non reconnaissable par aucune séquence d'acide nucléique connue, mais qui peut être construit en séquences de 10, 000 nucléotides ou plus. Cette longueur a été choisie pour réduire le nombre de contigs de matière noire étrangère (artefactuelle) tout en incluant la matière noire microbienne. Le GutFeelingKB est donc une base de connaissances complète sur le microbiome intestinal humain sain.
Cela a ensuite été utilisé comme référence pour construire un modèle de rapport standard où les microbiomes individuels peuvent être signalés, pour permettre une comparaison directe des résultats entre les études et les échantillons.
Les scientifiques ont également conçu un nouveau flux de travail à l'aide de plusieurs programmes informatiques et d'une base de données de séquences microbiennes filtrées de l'intestin appelée Filtered-nt, contenant près de 35, 000, 000 séquences permettant une interprétation biologiquement pertinente des séquences d'échantillons, tout en offrant l'assurance que l'ensemble de l'espace de séquence connu a été inclus.
Ainsi, GutFeelingKB représente une collection soigneusement organisée de séquences de nucléotides avec des métadonnées de 157 organismes de 60 genres.
Le microbiome humain sain contient ainsi des membres de 8 phylums, 18 familles, 60 genres et 109 espèces, principalement des phylums Firmicutes (40 %) et Bacteroidetes (20 %). 20% supplémentaires proviennent d'actinobactéries. Parmi les Firmicutes, plus de la moitié sont des Clostridia, suivi de près par Bacteroides, Bifidobactéries, Entérobactéries et Lactobacilles.
Tous les échantillons étaient positifs pour 84 des 109 organismes, qui représente peut-être la liste des espèces de base.
Cependant, il est important de noter que partout dans le monde, des organismes spécifiques qui ne figurent pas sur le GutFeelingKB ont été cartographiés, comme les Fusobactéries, certaines espèces Actinobacter et Bacteroides. La fonction de cette plateforme sera de servir de rampe de lancement pour comparer les résultats d'analyses d'échantillons d'individus sains, et de mieux comprendre les variations observées sous l'influence de facteurs alimentaires, maladies et médicaments.
Bacteroides est le genre le plus abondant dans de nombreux pays, en santé. Ceux-ci sont généralement bénéfiques à l'intérieur de l'intestin, mais s'ils s'échappent, ils saisissent l'occasion de provoquer des infections qui sont souvent résistantes aux médicaments et peuvent conduire à un taux de mortalité de 20 %. Cependant, dans l'intestin, ils protègent contre d'autres agents pathogènes et aident à décomposer les glucides dans l'alimentation.
De la même manière, Les bifidobactéries sont parmi les premiers colons de l'intestin, se trouvent souvent dans les probiotiques, et produisent l'important acétate d'acide gras à chaîne courte (SCFA) qui renforce la barrière épithéliale intestinale contre l'infection. Une souche de Bifidobacterium longum a été trouvé chez un individu d'un groupe de personnes ayant une durée de vie particulièrement longue en Chine.
Le nombre de Bifidobacterium augmente avec un apport protéique plus élevé, et surtout avec des protéines végétales. Les fibres solubles alimentaires favorisent également sa croissance. Akkermansia est lié aux graisses saturées et à l'acide linoléique, mais négativement associé aux acides gras polyinsaturés (AGPI). Bacteroides ovatus augmente en nombre avec l'augmentation de la prise alimentaire, l'obésité et le tour de taille. Ces exemples nous aident à comprendre comment ces chiffres peuvent être utilisés pour prendre des mesures de santé afin de corriger les déséquilibres du microbiome à l'avenir.
Le modèle de rapport publié dans la présente étude est destiné à remplacer les formats non standardisés utilisés par différents groupes commerciaux et de recherche, ce qui entrave leur interprétation et leur comparaison. Il convertit les données de recherche en un rapport clinique, aider à le rendre immédiatement exploitable.
FecalBiome a trois domaines, Échantillon, Patient et résultat, ressemblant à un rapport de panel métabolique. Il permet également aux collaborateurs de partager rapidement de nombreuses informations, lorsque la recherche se déroule dans un large éventail d'emplacements. Le seuil de déclaration peut être défini individuellement en fonction de l'objectif de l'étude. Il rapporte l'abondance, l'abondance moyenne et des informations sur les microbes présents.
Le présent rapport permet ainsi de relier la recherche sur les bactéries intestinales à des informations de santé compréhensibles, le rendant pertinent pour la pratique médicale et le patient. Il permet également une comparaison facile entre les études. Cela aidera les produits de remplacement intestinal à être examinés plus rapidement, et aider la médecine factuelle à progresser.
L'étude a été publiée le 11 septembre. 2019, dans PLOS UN .
 Comment fonctionne votre côlon et pourquoi les coloscopies sont nécessaires
Comment fonctionne votre côlon et pourquoi les coloscopies sont nécessaires
 Qu'est-ce que le cancer gastrique (estomac) ? Signes, symptômes, causes
Qu'est-ce que le cancer gastrique (estomac) ? Signes, symptômes, causes
 Gattex approuvé pour le syndrome de l'intestin court
Gattex approuvé pour le syndrome de l'intestin court
 Comment le gluten cause la maladie coeliaque
Comment le gluten cause la maladie coeliaque
 Comment savoir si vous avez la grippe intestinale ?
Comment savoir si vous avez la grippe intestinale ?
 Le paléo ou le SCD fonctionnera-t-il pour moi ?
Le paléo ou le SCD fonctionnera-t-il pour moi ?
 Est-ce que j'ai les symptômes du syndrome de l'intestin qui fuit ?
La question de savoir si vous souffrez du syndrome de lintestin qui fuit est importante. La santé gastro-entérologique implique le bon fonctionnement dun grand nombre de processus corporels, dont beau
Est-ce que j'ai les symptômes du syndrome de l'intestin qui fuit ?
La question de savoir si vous souffrez du syndrome de lintestin qui fuit est importante. La santé gastro-entérologique implique le bon fonctionnement dun grand nombre de processus corporels, dont beau
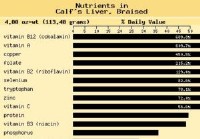 Recette SCD :steak de foie au citron et au poivre
Depuis quelques mois, je mintéresse de plus en plus aux abats. Jaimerais quavec le temps, je puisse compter sur la qualité des aliments que je mange pour obtenir toutes les vitamines et tous les minér
Recette SCD :steak de foie au citron et au poivre
Depuis quelques mois, je mintéresse de plus en plus aux abats. Jaimerais quavec le temps, je puisse compter sur la qualité des aliments que je mange pour obtenir toutes les vitamines et tous les minér
 Anévrisme de l'aorte abdominale
Ce que vous devez savoir sur les anévrismes de laorte abdominale Photo dun stent greffé pour réparer un anévrisme de laorte abdominale Quelle est la définition médicale de aortique anévrisme ? U
Anévrisme de l'aorte abdominale
Ce que vous devez savoir sur les anévrismes de laorte abdominale Photo dun stent greffé pour réparer un anévrisme de laorte abdominale Quelle est la définition médicale de aortique anévrisme ? U