il numero di copie del gene in tutto il genoma e l'analisi di espressione dei tumori gastrici primari e linee cellulari di cancro gastrico
Abstract
sfondo
cancro gastrico è una delle neoplasie più comuni in tutto il mondo e la seconda causa più comune di morte per cancro correlato. numero di copie del gene alterazioni svolgono un ruolo importante nello sviluppo del cancro gastrico e un cambiamento nel numero di copie del gene è uno dei principali meccanismi di una cellula tumorale per controllare l'espressione di potenziali oncogeni e geni oncosoppressori.
Metodi
per evidenziare i geni di potenziale rilevanza biologica e clinica nel carcinoma gastrico, abbiamo condotto una sistematica indagine basata su array di espressione genica e copiare i livelli di numero nei tumori gastrici primari e linee cellulari di cancro gastrico e ha convalidato i risultati utilizzando un'analisi trascrizione basato cattura affinità ( TRAC test) e in tempo reale qRT-PCR.
Risultati
analisi di microarray integrato hanno rivelato un totale di 256 geni che si trovavano nelle regioni ricorrenti di utili o perdite e aveva almeno 2 volte copia Number cambiamenti associati nella loro espressione genica. I livelli di espressione di 13 di questi geni, ALPK2
, ASAP1, CEACAM5
, CYP3A4, ENAH
, ERBB2
, HHIPL2
, LTB4R
, MMP9
, PERLD1
, PNMT
, PTPRA
, e OSMR
, sono stati validati in un totale di 118 campioni gastrici utilizzando sia la qRT-PCR o dosaggio TRAC. Tutti questi 13 geni sono stati differenzialmente espressi tra i campioni tumorali e tessuti non maligne (p < 0,05) e l'associazione tra numero e l'espressione genica di copia modifiche è stato convalidato per nove (69,2%) di questi geni (p < 0,05).
conclusione
In conclusione, l'espressione genica integrato e numero di copie analisi di microarray hanno evidenziato geni che possono essere di fondamentale importanza per la carcinogenesi gastrica. TRAC e qRT-PCR analisi convalidati i risultati di microarray e quindi il ruolo di questi geni come potenziali biomarcatori per il cancro gastrico.
Sfondo
A causa della mancanza di sintomi precoci adenocarcinoma gastrico è caratterizzato da una diagnosi tardivamente e le opzioni per insoddisfacenti trattamento curativo [1, 2]. Nonostante il calo della sua incidenza negli ultimi decenni, il cancro gastrico resta la seconda causa più comune di decessi correlati al cancro in tutto il mondo [3]. Circa il 90% di tutti i tumori gastrici sono adenocarcinomi derivanti dalla dell'epitelio [4]. Secondo la classificazione dei tumori gastrici di Lauren sono divisi in due principali sottotipi istologici, intestinali e diffondere [5].
Adenocarcinomi gastrici, come molti altri tumori solidi di origine epiteliale, sono spesso complesse in termini di integrità cromosomica [6, 7]. I tumori gastrici maligni sono noti per portare più aberrazioni nel loro genoma e tali alterazioni cromosomiche sono cruciali per l'attivazione e l'inattivazione di geni correlati al cancro [8-17]. Gene cambiamento numero di copie è uno dei principali meccanismi di una cellula tumorale per controllare l'espressione di geni cruciali per la sopravvivenza delle cellule e del cancro progressione [17-22]. Queste alterazioni del numero di copie comportano spesso un folto gruppo di geni che si trovano vicini l'uno all'altro nello stesso cromosoma. Per esempio; nei tumori gastrici regione 17q12-q21 spesso amplificata contiene geni, come ERBB2
, GRB7
, JUP
, PERLD1, PNMT
, PPP1R1B
, STARD3
, e TOP2A
[14, 17, 23]. Tuttavia, solo una minoranza di questi geni sono suscettibili di essere i veri geni del driver cancro che contribuiscono alla tumorigenesi, mentre altri possono essere amplificati semplicemente a causa della loro vicinanza cromosomica con i geni bersaglio di amplificazione [24, 25]. Un approccio di distinguere tali geni driver dal mutazioni passeggeri è quello di integrare numero di copie in tutto il genoma e dati di espressione, che permette l'identificazione di geni la cui trascrizione attivazione o la repressione è associato a un cambiamento del numero di copie in una cellula tumorale. Così, combinando le informazioni dal numero e di espressione genica di copia microarrays ad alta risoluzione, è possibile non solo per definire i punti di interruzione di copie cambiamenti numerici in grande dettaglio, ma anche per valutare il significato funzionale di questi cambiamenti e quindi forse identificare i geni che guidano il cancro insorgenza e la progressione.
Per evidenziare i geni potenziali come biomarcatori o obiettivi clinici nel carcinoma gastrico, abbiamo condotto una ricerca sistematica basata su serie ad alta risoluzione dei livelli del numero di copie e di espressione genica nei tessuti di cancro gastrico e linee cellulari. La nostra analisi basata su serie precedente ha mostrato che i guadagni di copia numero e le perdite di centinaia di geni sono associati con un contemporaneo aumento o diminuzione dell'espressione genica [17]. Nel presente studio, abbiamo aumentato la risoluzione dell'analisi numero di copie più di 20 volte di visualizzare con maggiore precisione i punti di interruzione delle alterazioni del numero di copie. Inoltre, abbiamo effettuato un'analisi trascrizionale di geni localizzati nelle regioni cromosomiche alterati per identificare i geni la cui deregolazione è associata con il fenotipo maligno.
Metodi
tessuti cancro gastrico e linee cellulari
Questo progetto di ricerca è stato rivisto e approvato dal Comitato Etico del Dipartimento di Genetica medica e Chirurgia e autorizzato dal Clinical Review Board di Helsinki University Central Hospital. campioni di tessuto gastrico sono stati raccolti prospetticamente da pazienti sottoposti a chirurgia gastrica o gastroscopia presso l'Ospedale Centrale Helsinki University tra il 1999 e il 2007. Il consenso informato è stato ottenuto da ciascun paziente partecipante. Tredici freschi tessuti tumorali congelati primari gastrici e sette linee di cellule di cancro gastrico sono stati scelti per l'analisi microarray (Tabella 1). Il materiale tessuto costituito da due differenti sottotipi istologici, intestinale (n = 9) e diffusa (n = 4) ed i tumori sono stati situati in due siti differenti dello stomaco, del corpus (n = 8) e l'antro (n = 5 ). Complessivamente 111 tessuti gastrici e 7 linee di cellule di cancro gastrico sono stati inclusi nella qRT-PCR e il saggio trascrizione basato cattura affinità (TRAC) analisi (file aggiuntivo 1: I parametri clinici). I campioni di tessuto consisteva di 43 e 68 nonmalignant tessuti gastrici cancerose ed entrambi i sottotipi istologici di cancro gastrico erano rappresentati (intestinale, n = 42; diffusa, n = 25; una delle sconosciuta istologia). campioni di tessuto gastrico sono stati conservati a -80 ° C. Per verificare la percentuale di tumore e istologico dei campioni, campioni congelati sono stati incorporati in Tissue-Tek ottobre Compound (Sakura Finetek, Torrance, CA, USA) e 5 micron di ghiaccio sezioni congelate sono stati preparati e macchiati con Trypan Blue. Istologia dei campioni di cancro gastrico è stata valutata da un esperto patologo (M.-L. K.-L.). Tissue-Tek è stato rimosso dai tessuti prima di acido nucleico extractions.Table 1 parametri clinici per i campioni analizzati su array di ibridazione genomica comparativa (CGH) e microarray di espressione.
primari gastrico tumors
Age/sex
Histology
Location
14TA
58/M
Intestinal
Corpus
200A
57/F
Intestinal
Corpus
222A
50/M
Intestinal
Corpus
232A
83/M
Intestinal
Corpus
3TC
57/F
Intestinal
Corpus
4T/N
72/M
Intestinal
Corpus
10TB
59/M
Intestinal
Antrum
17TA
77/M
Intestinal
Antrum
185B
78/F
Intestinal
Antrum
1AT/N
41/F
Diffuse
Corpus
6TB
77/F
Diffuse
Corpus
9TD
74/F
Diffuse
Antrum
13TA
56/F
Diffuse
Antrum
linee cellulari di carcinoma gastrico
Età /sesso
Istologia
Origine
AGS
54 /F
adeno-carcinoma
tumore primitivo
KATOIII
55 /M
Diffuse
versamento pleurico
MKN-1
72 /M
carcinoma adeno-squamose
metastasi linfonodali
MKN-7
39 /M
intestinale
metastasi linfonodali
MKN-28
70 /F
intestinale
metastasi linfonodali
MKN-45
62 /F
Diffondere
metastasi epatiche
TMK-1
21 /M
Diffuse
linfa metastasi nodo
linee cellulari AGS e KATOIII sono stati ottenuti da American Type Culture Collection (Rockville, MD, USA) e MKN-1, MKN-7 , MKN-28, MKN-45, e linee cellulari TMK-1 erano un gentile dono da Hiroshi Yokozaki, Kobe University Graduate School of Medicine, Kobe, in Giappone [26]. cellule AGS sono state coltivate in mezzo F12 di Kaighn (2 mM glutammina, 10% FBS, 100 U /ml di penicillina-streptomicina), cellule KATOIII in mezzo IMDM (2 mM glutammina, 10% FBS, 100 U /ml di penicillina-streptomicina) e tutti altre linee cellulari in terreno RPMI-1640 (10% FCS, 2 mM glutammina, 100 U /ml di penicillina-streptomicina). Tutte le cellule sono state coltivate a 37 ° C e 5% di CO
2.
RNA e l'estrazione del DNA
Prima di RNA e DNA estrazioni, il tessuto congelato è stato immerso in reagente RNAlater-ICE (Ambion, Austin, TX , USA) e conservati a -80 ° C per 16 ore per stabilizzare l'RNA. La metà del campione di tessuto (~ 25 mg) è stato omogeneizzato in tampone RLT-β-mercaptoetanolo lisi (RNeasy Mini Kit, Qiagen Inc., Hilden, Germania) e l'altra metà in ATL-buffer (DNeasy Sangue e Kit Tissue, Qiagen) utilizzando l'omogeneizzatore Ultra-Turrax (IKA Works, Wilmington, NC, USA). RNA è stato estratto utilizzando il kit RNeasy mini, compreso il trattamento DNasi opzionale, e il DNA utilizzando il DNeasy Sangue e Kit Tissue. Per le linee di cellule di cancro gastrico, 1 × 10 7 cellule sono state lisate con una siringa e l'ago nel tampone di lisi o RLT-β-mercaptoetanolo o ATL-buffer prima di RNA e DNA estrazioni, rispettivamente. le concentrazioni di RNA e DNA sono stati misurati utilizzando NanoDrop1000 (Thermo Fisher Scientific, Waltham, MA, USA) e la qualità dell'RNA è stata valutata utilizzando di Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). Solo RNA che mostrano distinte 18S e 28S ribosomiale picchi nell'analisi Bioanalyzer e 260/280 indici riportati sopra 2.0 sono stati accettati per ulteriori analisi. Analizza
Array CGH e l'espressione genica microarray
Tredici tumori gastrici e sette linee di cellule di cancro gastrico sono stati analizzati sulle oligoarrays 244K Human Genome CGH (G4411B, Agilent Technologies). Tre dei tumori e tutte le sette linee di cellule sono stati analizzati anche utilizzando l'espressione genica oligoarrays 44K intero genoma umano (G4112F, Agilent Technologies) (Figura 1). I 260/280 rapporti medi per questi campioni erano 2.1 per RNA e 1.8 per DNA, e tutti i campioni di RNA avuto chiari picchi 18S e 28S ribosomiale nell'analisi Bioanalyzer indicante buona qualità (dati non mostrati). Array CGH esperimenti sono stati effettuati utilizzando kit Human Genome CGH microarray 244A (Agilent Technologies). L'etichettatura e l'ibridazione sono stati eseguiti secondo il protocollo di Agilent (v5.0, giugno 2007). In breve, 1,5 mg di DNA del campione e 1,5 mg di DNA di riferimento del sesso-abbinato (Essere umano DNA genomico, Promega, Madison, WI, USA) erano doppio digerito con AluI
e RSAI
enzimi di restrizione (Promega). Il DNA digerito è stato etichettato con Agilent DNA genomico Labeling Kit plus. DNA campione è stato etichettato con Cy5-dUTP e il DNA di riferimento con Cy3-dUTP, rispettivamente. Labeled DNA è stato purificato con microcontrollore YM-30 filtri (Millipore, Billerica, MA, USA). A seguito dei DNA di purificazione, di esempio e di riferimento sono stati raggruppati e ibridati alla matrice con 50 mg di Human Cot-1 DNA (Invitrogen, Carlsbad, CA, USA) a 65 ° C, 20 rpm per 40 h. L'ibridazione è stata eseguita con il kit Agilent Oligo aCGH ibridazione. Prima di scansione, i vetrini sono stati lavati secondo il protocollo. Oltre alle ibridazioni DNA campione sopra descritte, riferimento maschio DNA (Cy3) è stato ibridato contro riferimento DNA femminile (Cy5) secondo il medesimo protocollo da utilizzare come una matrice di riferimento per l'analisi dei dati aCGH. Figura 1 Diagramma di flusso che descrive diverse fasi dello studio. Esperimenti di espressione genica
sono state eseguite utilizzando l'intero kit Genoma Umano oligo microarray (Agilent Technologies), e l'etichettatura e l'ibridazione secondo il protocollo Agilent (quella 5.7, marzo 2008). In breve, 2 mg di RNA totale del campione e RNA di riferimento (un pool di linee cellulari di cancro 10, non-gastrico, ATCC, Manassas, MA, USA) sono stati etichettati con l'Amp Labeling Kit Agilent rapida. RNA campione è stato etichettato con Cy5-dCTP e il DNA di riferimento con Cy3-dCTP, rispettivamente. RNA etichettati è stato poi purificato mediante RNeasy mini colonne di rotazione (Qiagen). Ibridazione è stata eseguita con Agilent Gene Expression Kit ibridazione e campioni sono stati ibridati a 65 ° C, 10 rpm per 17 h e lavato secondo il protocollo prima della scansione. Entrambi gli scivoli microarray aCGH e di espressione genica sono stati sottoposti a scansione utilizzando il DNA microarray scanner (Agilent Technologies) e analizzati con il software Feature Extraction (v9.5.1.1.).
Numero di copie ad alta risoluzione profilazione
Tutti i dati del numero di copie è disponibile all'indirizzo http: //www. cangem org (numero di accesso: CG-EXP-49). [27]. CGH software Analytics Agilent (v3.5.14) è stata applicata per identificare i cambiamenti del numero di copie. dati microarray è stata la qualità filtrato utilizzando le informazioni outlier ottenuto dall'analisi Feature Extraction. Sonde indicati come valori anomali sono stati rimossi da ulteriori analisi. Inoltre, sono stati applicati i seguenti filtri di aberrazione: numero minimo di sonde della regione = 3, minimo assoluto di registro 2 Rapporto per regione = 0,27, e il numero massimo di regioni aberranti = 1000. Il registro 2 rapporto di 0,27 corrisponde ad una variazione di 1,2 volte nel numero di copie. In CGH Analytics, ogni rapporto aCGH è stato convertito in un registro rapporto 2 seguito da un Z-normalizzazione. Il maschio vs. matrice riferimento femmina è stato usato come un array calibrazione nell'analisi dei dati. A causa delle differenze di genere tra gli array che potrebbero causare distorsione nell'analisi, cromosomi X e Y sono stati esclusi dalla taratura. Algoritmo ADM-2 con un livello di soglia di 12,0 è stato usato per identificare il numero di copie di geni alterazioni in singoli campioni e linee cellulari. Minimal regioni comuni della alterazione delle 20 campioni sono stati calcolati, compresa la posizione cromosomica dimensioni e della modifica in paia di basi. Una aberrazione è stata definita come ricorrente, se fosse presente in almeno il 25% dei campioni (Tabella 2) .table 2 Minimal regioni comuni di recidiva (≥25%) copia alterazioni numero.
Alterazione
tessuti (n = 13)
linee cellulari (n = 7)
Frequenza
Size (Mb)
posizione (Mb)
possibili geni bersaglio
+ 1q41-q43.1
2 3
25 %
17.30
216,31-233,61
ENAH, AGT, CAPN2, LEFTY2, LGALS8
+ 5p13.3-q11.1
1 4
25%
19.41
30,18-49,60
OSMR, RNASEN
+ 7q21.3-q22.1
4 3
35%
4,60
97,33-101,93
CYP3A4, AZGP1, VGF
+ 8q24.13-q24.3 3
2
25%
19,8
126,45-146,25
ASAP1, BAI1, KHDRBS3
+ 8q24.3
6 3
45%
2.23
143,59-145,82
GML, LYPD,
AK3
+ 14q11.2
0
5
25%
1,05
22,89-23,94
LTB4R
+ 17q12-q21.1
3 3
30%
0,28
35,02-35,30
ERBB2, PPP1R1B, PERLD1, PNMT
+ 17q22-q24.2 2
3
25%
13.65
50,45-64,10
AXIN2, RNF43
+ 19q12-qter
4 3
35%
29.36
33,89-63,25
CEACAM5, APOC1, APOE, CEACAM7, FTL, FUT1, GPR4, NPD, KCNN4, KLK1, KLK12, LYPD3, NLRP7, CCNE1
+ 20p13-qter
5
3
40%
57,94
0,04-57,98
PTPRA, BLCAP, CD40, CHGB, CSt3, EYA2, PI3, ID1, MMP9, BMP7
-9p24.3-p21 .1
3 4
35%
27.81
1,05-28,86
MTAP, CD274, INSL4, JAK2, MLANA, SMARC2, TUSC1
-18q12. 3-q22.2 3
5
40%
26.11
39,48-65,59
SMAD7, SERPINB2 /B3 /B4 /B5
-18q22.3 -qter 2
5
35%
3.69
70,95-74,65
TSHZ1
-21q11.2-q21.1 3
3
30%
4,07
14,37-19,44
HSPA13
-Xq28 4
1
25%
1.21
152.24- 153.45
-
Numero di casi, le dimensioni minime delle regioni comuni (Mb), e la posizione cromosomica dell'alterazione (Mb) sono indicati come pure possibili geni bersaglio. regioni CNV non sono riportati nella tabella. . Numero di copie del guadagno (+), la perdita del numero di copie (-)
Analisi dell'espressione genica microarray
Tutti i dati di espressione genica è disponibile all'indirizzo http: //www cangem org (numero di accesso: CG-EXP.. -49) [27]. i risultati sono stati microarray qualità filtrati utilizzando valori anomali definiti dal software Feature Extraction e normalizzati in base al metodo Loess, che era inclusa nel pacchetto software. L'analisi di espressione genica è stata limitata ai geni situati nelle regioni cromosomiche con aberrazioni ricorrenti (Tabella 2). L'obiettivo di questo approccio è stato quello di mettere in evidenza i cambiamenti di espressione genica che sono stati associati con i cambiamenti nel numero di copie del gene, e potrebbe quindi rappresentare potenziali oncogeni o geni oncosoppressori con un ruolo funzionale nel cancro. Innanzitutto, una mediana rapporto log10 espressione è stato calcolato per tutte le sonde di targeting lo stesso gene. Poi, in due analisi separate per gli utili e le perdite, il livello di espressione mediana di ciascun gene è stata confrontata tra i campioni con copia numero di guadagno /perdita e campioni con normale numero di copie per valutare l'effetto delle alterazioni del numero di copie sull'espressione genica. espressione genica piega modifiche (FC) sono stati calcolati sia dividendo l'espressione mediana dei campioni cancerosi dall'espressione mediana dei campioni non maligne o dividendo l'espressione mediana dei campioni tumorali con alterazioni del numero di copie (G1) con l'espressione mediana dei campioni di cancro con normale numero di copie (G0). Almeno un cambio di 2 volte numero di copie associato di espressione genica è stato considerato significativo. Sulla base di questi dati, 13 geni ALPK2
, ASAP1
, CEACAM5
, CYP3A4
, ENAH
, ERBB2, HHIPL2, LTB4R
, MMP9, OSMR
, PERLD1
, PNMT
, e PTPRA
, sono stati scelti per essere ulteriormente validati con analisi qRT-PCR e TRAC (analisi trascrizione con l'aiuto di cattura affinità) test. I risultati dell'analisi microarray integrato sono stati confrontati con tre studi pubblicati in precedenza che si integrano in modo sistematico del numero di copie e di espressione genica dei dati a livello di genoma [15-17]. Real-time qRT-PCR Real-time qRT- PCR è stata effettuata per 2 geni, ALPK2
(18q21.31-q21.32) e HHIPL2
(1q41). I livelli di espressione sono stati misurati in 82 tessuti gastrici (46 cancerose e 36 tessuti non maligne) e in 7 linee di cellule di cancro gastrico (file aggiuntivo 1: parametri clinici). 1 mg di RNA totale è stato convertito in cDNA usando virus della leucemia murina Moloney-trascrittasi inversa (Promega, Madison, WI, USA) e primer casuali (Invitrogen) in un volume di 50 ml per 1 ora a 37 ° C. La reazione era inattivato al calore (95 ° C, 3 min) e riempito ad un volume finale di 200 microlitri con acqua grado molecolare. Le trascrizioni sono state quantificate utilizzando i prodotti di espressione genica Assays-on-DemandTM (Hs01085414_m1 per ALPK2
e Hs00226924_m1 per HHIPL2
) secondo il protocollo del produttore (Applied Biosystems, Foster City, CA, USA). Tutti i primer erano situati sui confini esone-esone. In breve, 2 ml di cDNA è stato mescolato con 1,25 ml di primer e sonde specifiche marcate con colorante FAM-giornalista. 12,5 ml di TaqMan ® Universal PCR Mastermix e acqua RNase-free sono stati aggiunti a un volume totale di 25 ml. Umana 18S rRNA servito come controllo endogeno per normalizzare i livelli di espressione nella successiva analisi quantitativa. La sonda 18S è stato marcato con colorante VIC-giornalista per consentire PCR multiplex con i geni bersaglio. Le condizioni di PCR erano le seguenti: 50 ° C per 2 min, 95 ° C per 10 min, seguiti da 40 cicli di 95 ° C per 15 s e 60 ° C per 1 min. Ogni campione è stata misurata in triplicato ed i dati sono stati analizzati con il metodo delta-delta per confrontare i risultati di espressione relativi (2 - [Controllo Ct campione-Ct])
TRAC saggio
analisi Trascrizione con l'aiuto di. cattura affinità (TRAC) saggio [28] è stata eseguita per 11 diversi geni in 88 tessuti gastrici (53 cancerose e 35 tessuti non maligni) e 7 linee di cellule di cancro gastrico (file aggiuntivo 1: parametri clinici). I geni inclusi nell'analisi sono stati ENAH
(1q42.12), OSMR
(5p13.1), CYP3A4
(7q21.1) ASAP1
(8q24.1-q24.2), LTB4R
(14q11.2-q12), PERLD1
(17q12), ERBB2
(17q21.1), PNMT
(17q21-q22), CEACAM5
(19q13.1-q13 .2), PTPRA
(20p13), e MMP9
(20q11.2-q13.1). Il vantaggio del test TRAC è che i livelli di espressione di geni multipli possono essere misurate simultaneamente da un singolo campione riducendo così la quantità di RNA campione necessario per l'analisi. Ciò è particolarmente importante per l'analisi di campioni di tessuto spesso scarse clinici.
Analisi TRAC è stata eseguita a Plexpress (Helsinki, Finlandia). reagenti personalizzati TRACPackTM di mRNA (Plexpress) sono stati utilizzati per l'analisi. In breve, 90 ml di ibridazione Mix (contenente etichettati sonde di rilevamento gene-specifici e biotinilati sonde oligo-DT) per pozzo è stato dispensati ad una piastra a 96 pozzetti PCR. Due microgrammi di RNA campione è stato applicato a ciascun pozzetto in un volume di reazione totale di 100 microlitri. Un importo pari (30 Amol /reazione) del singolo filamento di controllo oligonucleotide ibridazione sintetico 62-mer, tra cui un poli-Una coda, è stato aggiunto ad ogni campione prima di ibridazione. L'ibridazione è stata effettuata a 60 ° C, 650 giri al minuto per 120 minuti (Thermomixer comfort, Eppendorf, Amburgo, Germania). Dopo la cattura ibridazione affinità, la purificazione, e eluizione sono stati fatti usando il martin pescatore Flex (Thermo Fisher Scientific, Vantaa, Finlandia) del processore di particelle magnetiche. perline TRACPACK ™ magnetiche streptavidina accoppiata (50 ug, Plexpress) sono stati aggiunti alla miscela di ibridazione e permesso di legarsi biotinilato mRNA-probe-oligo (dT) -hybrids per 30 minuti, dopo di che le perle sono state lavate 5 volte con wash tampone per rimuovere il materiale non legato. sonde specifiche per RNA marcate sono state eluite con tampone di eluizione e rilevati da elettroforesi capillare, utilizzando il sequencer ABI3100 (Applied Biosystems, Cheshire, UK). I dati sono stati analizzati utilizzando il software TRACParser (Plexpress).
L'analisi statistica dei dati qRT-PCR
Un test non parametrico di Mann-Whitney per due campioni indipendenti è stato applicato per determinare la significatività statistica delle differenze nell'espressione di mRNA relativa livelli di ALPK2
e HHIPL2
in campioni gastrici non maligne e cancerose, nonché in campioni di cancro gastrico di diversi sottotipi istologici o TNM stadi. Un valore di p < 0.05 è stato considerato statisticamente significativo (SPSS 17.0). Inoltre, in due analisi separate per i guadagni e le perdite, i livelli di espressione nei campioni tumorali con copia guadagni numero o le perdite (G1) sono stati confrontati con campioni tumorali con normale numero di copie (G0) per valutare l'associazione tra numero di copie e l'espressione genica. i dati del numero di copie erano disponibili per 37 dei campioni gastrici inclusi nell'analisi qRT-PCR (sui file 1: I parametri clinici). espressione genica piega cambiamenti sono stati calcolati dividendo l'espressione media di un gruppo (ad esempio campioni di cancro) con l'espressione significa dell'altro gruppo (ad esempio, i campioni non maligne).
L'analisi statistica dei dati di analisi TRAC
Un controllo ibridazione sintetico è stato utilizzato nel normalizzazione dei dati per rimuovere eventuali variazioni non biologica nei dati. Per ogni obiettivo, il segnale intensità relative a questo controllo ibridazione interno sono stati calcolati. Per i campioni di tessuto nove analizzati in replica significa intensità del segnale è stato utilizzato. Un test non parametrico di Mann-Whitney per due campioni indipendenti è stata applicata per determinare la significatività statistica delle differenze tra i relativi livelli di espressione di mRNA di ASAP1
, CEACAM5
, CYP3A4
, ENAH
, ERBB2, LTB4R
, MMP9, OSMR
, PERLD1
, PNMT
, e PTPRA
in campioni gastrici non maligne e cancerose, nonché in campioni di cancro gastrico di diversi sottotipi istologici o TNM stadi. Un valore di p < 0.05 è stato considerato statisticamente significativo (SPSS 17.0). Il confronto dei livelli di espressione genica in campioni con e senza numero di copia alterazioni è stata eseguita come è stato descritto in precedenza per il qRT-PCR. . I dati del numero di copie erano disponibili per 43 campioni di cancro gastrico inclusi nell'analisi saggio TRAC
Risultati
copia del gene numero aberrazioni in tutte le nazioni il numero di copie del gene cambia in singoli campioni sono riportati nel file aggiuntivo 2: numero di copie modifiche rilevato da analisi aCGH. Minimal regioni comuni ricorrenti (≥25%) alterazioni nonché le loro dimensioni, frequenza, possibili geni bersaglio, e la posizione cromosomica in paia di basi sono mostrati in Tabella 2. La ricorrente acquisita regioni erano situati 1q41-q43.1 (25% ), 5p13.3-q11.1 (25%), 7q21.3-q22.1 (35%), 8q24.13-q24.3 (25%), 8q24.3 (45%), 14q11.2 ( 25%), 17q12-q21.1 (30%), 17q22-q24.2 (25%), 19q12-qter (35%), e 20p13-qter (40%). Le regioni ricorrenti soppresso erano situati 9p24.3-p21.1 (25%), 18q12.3-q22.2 (40%), 18q22.3-qter (35%), 21q11.2-q21.1 (30 %), e Xq28 (25%). Tutti del numero di copie variazioni ricorrenti erano rilevabili sia in tumori gastrici primarie e linee cellulari di cancro gastrico, tranne per il 14q11.2, che è stato modificato solo in cinque linee cellulari.
Numero Copy genica associata cambia
Complessivamente 256 individuale geni (10% di tutti i geni situati nelle regioni cromosomiche ricorrenti con alterazioni del numero di copie) hanno mostrato almeno un numero di copie di 2 volte il cambiamento associato nella loro espressione (range 2,0-34,6, mediana 3,8) (sui file 3: copia il numero associato gene cambiamenti di espressione). 226 di questi geni sono stati overexpressed e situati in regioni ricorrenti di numero di copia guadagni, mentre i 30 geni sono stati underexpressed e situate in regioni ricorrenti di perdite del numero di copie. fold change di espressione genica è stata calcolata confrontando i livelli di espressione di campioni con alterazioni del numero di copie di campioni con il normale numero di copie in un dato gene. Pertanto, una variazione positiva volte si riferisce a un aumento del numero di copie di guadagno legate nell'espressione genica, mentre una variazione negativa volte si riferisce a una diminuzione di perdita di numero di copie relative nell'espressione genica.
HHIPL2
(HHIP-simile 2) gene, amplificato nella regione 1q41-q43.1, ha mostrato il più alto numero di copie guadagno sovraespressione associata a cancro gastrico secondo l'analisi di microarray integrato (FC = 26,9). In generale, la più alta espressione del gene piegare le modifiche tra i campioni tumorali con e senza numero di copia guadagni sono stati rilevati alla regione 19q in quanto dei 40 geni che mostrano > si trovavano del numero di copie di 5 volte associata cambiamenti nella loro espressione, 19 (47,5%) nei 19q regione (file aggiuntivo 3: cambiamenti Copia numero associato gene di espressione). Il gene più underexpressed nelle regioni ricorrenti di copia perdite numero era ALPK2
(alfa-chinasi 2) (FC = -34,6) situato a 18q12.3-q22.2.
In precedenza, tre studi da noi e gli altri sono stati pubblicati che si integrano in modo sistematico del numero di copie e di espressione genica dei dati a livello di genoma per identificare i geni la cui espressione è cambiata a causa di un numero di copie alterazione cancro gastrico [15-17]. Il confronto dei geni sovrapposizione tra questi studi e l'attuale studio ha rivelato 20 geni TOMM20
(1q42.3), GGPS1
(1q43), CYP3A4
(7q21.1), MTAP
(9q21 .3), ASAP1
(8q24.1-q24.2), PPP1R1B
(17q12), ERBB2
(17q12-q21), SERPINB3
(18q21.3), SERPINB8
(18q21.3), WDR7
(18q21.2-Q22), HIF3A
(19q13.32), ZNF480
(19q13.33), IL4I1
(19q13.3-q13.4 ), CSt3
(20p11.21), PTPRA
(20p13), SLC13A3
(20q12-q13.1), DDX27
(20q13.13), PARD6B
(20q13.13 ), SGK2
(20q13.2), e TUBB1
(20q13.32) che sono stati sia acquisita e overexpressed o cancellato e underexpressed nel nostro studio e in almeno uno degli studi pubblicati in precedenza. Precedentemente pubblicato i dati insieme con i risultati attuali forniscono un'ulteriore prova del ruolo biologico di questi geni nel cancro gastrico.
Convalida di potenziali geni bersaglio cancro gastrico Real-time qRT-PCR ha mostrato che l'espressione di HHIPL2
è stato 7.4 volte più elevata nei campioni di cancro gastrico rispetto ai tessuti gastrici non maligne (p < 0,05). Inoltre, la sovraespressione di HHILP2
era significativamente associato con un aumento del numero di copie (p < 0,05) come l'espressione di HHIPL2
era 17,4 volte maggiore nei campioni tumorali con numero di copie guadagno di HHIPL2
(g1 ) che nei campioni tumorali con normale numero di copie di questo gene (G0) (Tabelle 3 e 4). Secondo l'analisi qRT-PCR c'era una sottoespressione 2,9 volte di ALPK2
nei tumori gastrici con perdite del numero di copie (G1) rispetto a tumori gastrici con normale numero di copie di ALPK2
(G0) (p < 0,05 ). Sorprendentemente, però, l'espressione di ALPK2
in tumori gastrici, in generale, è stato di 1,9 volte superiore (p < 0,05) che nei tessuti gastrici non maligne (Tabelle 3 e 4). sottotipo istologico o TNM stadi non hanno avuto un effetto statisticamente significativo sulla espressione di HHIPL2
o ALPK2
(Tabella 3) .table 3 I risultati del test non parametrico di Mann-Whitney per la qRT-PCR e analisi dei dati TRAC (SPSS17.0).
Gene
cromosoma
Cancer vs. non maligne
intestinale vs diffuso
G1 vs G0
M0 vs M1
T1-2 T3-4 vs.
N0 vs N1-3
ALPK2
18q21.31-q21.32 p < 0.05
p = 0,104
p
< 0.05
p = 0,451
p = 0,072
p = 0,378
ASAP1
8q24.1-q24.2
p < 0.001
p = 0,319
p = 0,396
p = 0,208
p = 0,232
p = 0,289
CEACAM5
19q13.1-q13.2
p < 0.001
p = 0,061
p = 0,254
p = 0,543
p = 0,197
p = 0,253
CYP3A4
7q21.1
p <
 Malattie digestive al secondo sguardo
Malattie digestive al secondo sguardo
 La gastroenterologia di San Francisco presenta il nostro portale per i pazienti
La gastroenterologia di San Francisco presenta il nostro portale per i pazienti
 Vantaggi di una dieta blanda per la gestione dei sintomi
Vantaggi di una dieta blanda per la gestione dei sintomi
 Perché sottoporsi a una colonscopia?
Perché sottoporsi a una colonscopia?
 Le 25 migliori ricette di Pasqua al cioccolato SIBO
Le 25 migliori ricette di Pasqua al cioccolato SIBO
 Come due ragazzi del Michigan hanno iniziato un insolito esperimento di consulenza sanitaria
Come due ragazzi del Michigan hanno iniziato un insolito esperimento di consulenza sanitaria
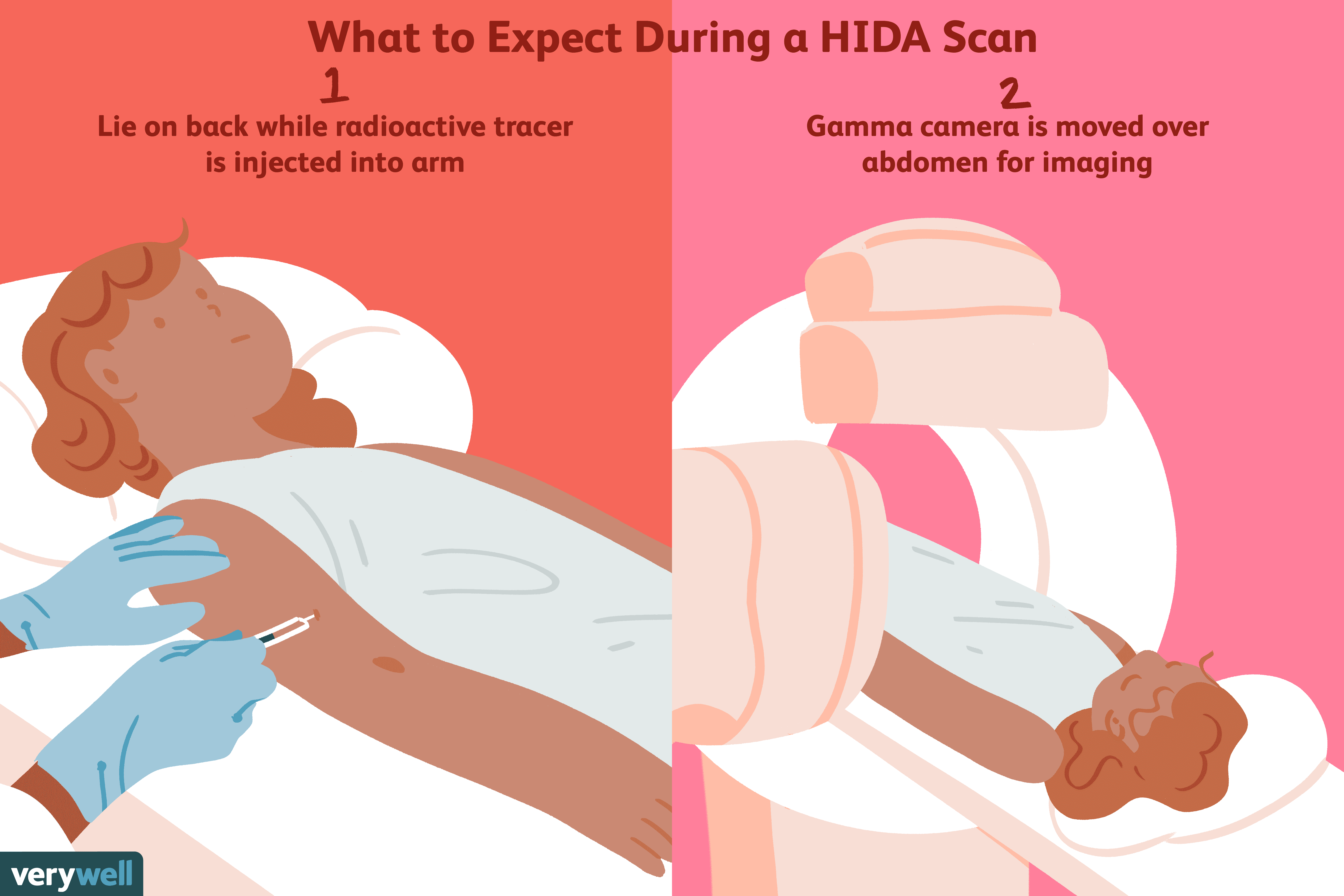 Che cos'è una scansione dell'acido epatobiliare imminodiacetico (HIDA)?
Una scansione dellacido iminodiacetico epatobiliare (HIDA), che a volte è anche chiamata colescintigrafia o scintigrafia epatobiliare, è un tipo di test di imaging nucleare che viene eseguito per visu
Che cos'è una scansione dell'acido epatobiliare imminodiacetico (HIDA)?
Una scansione dellacido iminodiacetico epatobiliare (HIDA), che a volte è anche chiamata colescintigrafia o scintigrafia epatobiliare, è un tipo di test di imaging nucleare che viene eseguito per visu
 Gli uomini che mangiano yogurt due volte a settimana hanno meno probabilità di sviluppare il cancro all'intestino
Una nuova ricerca ha rivelato che gli uomini che mangiano due o più porzioni di yogurt alla settimana possono ridurre il rischio di sviluppare escrescenze precancerose che possono portare al cancro de
Gli uomini che mangiano yogurt due volte a settimana hanno meno probabilità di sviluppare il cancro all'intestino
Una nuova ricerca ha rivelato che gli uomini che mangiano due o più porzioni di yogurt alla settimana possono ridurre il rischio di sviluppare escrescenze precancerose che possono portare al cancro de
 Tylenol (acetaminofene) danni al fegato
Dati sul danno epatico del paracetamolo (Tylenol) Tylenol è un agente analgesico (analgesico) e antipiretico molto efficace. Tuttavia, anche lassunzione di troppo Tylenol (un sovradosaggio) può cau
Tylenol (acetaminofene) danni al fegato
Dati sul danno epatico del paracetamolo (Tylenol) Tylenol è un agente analgesico (analgesico) e antipiretico molto efficace. Tuttavia, anche lassunzione di troppo Tylenol (un sovradosaggio) può cau