 Personer med ubehandlet Wilsons sykdom kan ha en forventet levetid på 40 år; tidlig diagnose og behandling kan imidlertid øke levetiden.
Personer med ubehandlet Wilsons sykdom kan ha en forventet levetid på 40 år; tidlig diagnose og behandling kan imidlertid øke levetiden. Wilsons sykdom er en svært sjelden genetisk lidelse arvet i et autosomalt recessivt mønster som kan overføres til neste generasjon fra foreldre som bærer en eller begge kopier av det berørte genet. Det er anslått at 1 av 40 000 mennesker over hele verden er rammet av Wilsons sykdom.
For å utvikle Wilsons sykdom må du arve to kopier av det defekte genet, en fra hver forelder. Hvis du bare arver én kopi av det defekte genet, vil du være bærer og gi genet videre til neste generasjon, men du vil ikke utvikle sykdommen.
Vanligvis utvikler symptomer på Wilsons sykdom mellom 12 og 23 år, og ubehandlede personer kan ha en forventet levealder på 40 år . Imidlertid kan tidlig diagnose, etterfulgt av riktig behandling, øke levetiden.
Wilsons sykdom er forårsaket av en defekt i ATP7B genet, som forhindrer metabolisering og eliminering av kobber fra kroppen, og kan potensielt utvikle seg til en livstruende tilstand.
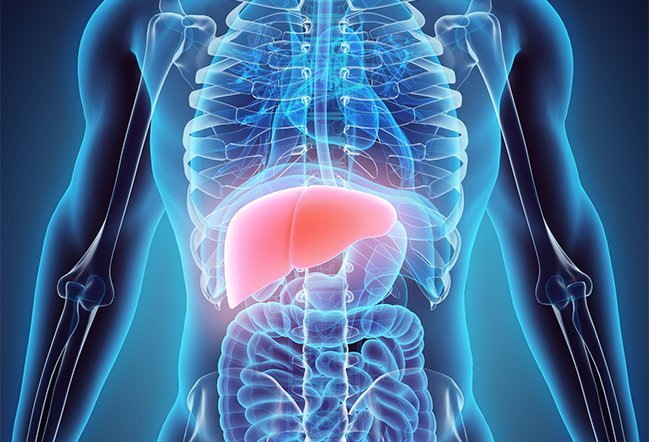
Selv om kroppen din trenger en liten mengde kobber for visse kroppsfunksjoner, er en overflødig mengde av dette mineralet giftig og kan føre til komplikasjoner. Leveren spiller en viktig rolle i metabolismen av kobber med overflødig kobber overført til gallen, som senere elimineres gjennom avføring.
Wilsons sykdom påvirker ulike organer, noe som resulterer i ulike symptomer som kan omfatte:
Diagnosen av Wilsons sykdom gjøres ikke lett i de innledende stadiene fordi symptomene som forårsakes er korrelert med mange andre medisinske tilstander, spesielt med tilstander som involverer lever og nerver.
Leger diagnostiserer Wilsons sykdom ved følgende:
Leger utfører magnetisk resonansavbildning og datatomografi for å oppdage eventuelle hjerneabnormiteter. Selv om disse funnene ikke hjelper med den første diagnosen av sykdommen, vil de bidra til å bekrefte den og bestemme det nåværende stadiet.
Leger kan utføre en leverbiopsi for å bestemme omfanget av kobberavsetning og mulig leverskade.
Leger gjennomfører en prosedyre der de trekker ut en liten del av levervevet ved å bruke en nål. Denne prosedyren utføres under påvirkning av anestesi. Leger studerer det ekstraherte levervevet under et mikroskop for å se etter kobberavleiringer.
Wilsons sykdom kan ikke forebygges fordi den overføres gjennom gener fra foreldre uten kjent kur. Du kan imidlertid bremse utviklingen av sykdommen ved tidlig diagnose og behandling av symptomene.
Behandlingsalternativer for Wilsons sykdom inkluderer:
 Er dietten nok til å fikse fordøyelsesproblemer?
Tenk deg at det er 22. januar 1930 og du er en del av gravemannskapet som graver fundamentet til Empire State Building – den planlagte 102-etasjers skyskraperen designet for å være verdens høyeste byg
Er dietten nok til å fikse fordøyelsesproblemer?
Tenk deg at det er 22. januar 1930 og du er en del av gravemannskapet som graver fundamentet til Empire State Building – den planlagte 102-etasjers skyskraperen designet for å være verdens høyeste byg
 Begynner all sykdom i tarmen?
Hvorfor er vi så lidenskapelig opptatt av Leaky Gut Awareness? Poenget er dette:å fikse tarmen vil påvirke omtrent alle andre områder av kroppen din. Det er millioner av mennesker i verden som slite
Begynner all sykdom i tarmen?
Hvorfor er vi så lidenskapelig opptatt av Leaky Gut Awareness? Poenget er dette:å fikse tarmen vil påvirke omtrent alle andre områder av kroppen din. Det er millioner av mennesker i verden som slite
 Urinveisinfeksjon (UTI) Symptomer, diagnose, medisiner
Symptomer på urinveisinfeksjon (UTI) Symptomer på urinveisinfeksjon (UTI) kan være skremmende og en grunn til bekymring. Noen mennesker som har en UVI har ingen symptomer. Noen ganger kan en lege di
Urinveisinfeksjon (UTI) Symptomer, diagnose, medisiner
Symptomer på urinveisinfeksjon (UTI) Symptomer på urinveisinfeksjon (UTI) kan være skremmende og en grunn til bekymring. Noen mennesker som har en UVI har ingen symptomer. Noen ganger kan en lege di