Ein neuartiges Berechnungsverfahren zur Vorhersage von Proteinen in den Urin ausgeschieden wird vorgestellt. Das Verfahren basiert auf der Identifizierung einer Liste von Unterscheidungsmerkmale zwischen Proteinen als im Urin von gesunden Menschen und Proteine nicht gefunden Urinausscheidungs sein. Diese Funktionen werden verwendet, um einen Klassifikator zu trainieren, um die zwei Klassen von Proteinen zu unterscheiden. Wenn sie in Verbindung mit Informationen verwendet, von denen Proteine in erkrankten Geweben eines bestimmten Typs differentiell exprimiert werden gegen Citation:. Hong CS, Cui J, Ni Z, Su Y, Puett D, Li F, et al. (2011) A Computational Methode zur Vorhersage von Excretory Proteine und Anwendung auf die Identifizierung von Magenkrebs-Marker im Urin. PLoS ONE 6 (2): e16875. doi: 10.1371 /journal.pone.0016875 Herausgeber: Vladimir Brusic, Dana-Farber Cancer Institute, Vereinigte Staaten von Amerika Empfangen: 22. September 2010; Akzeptiert: 31. Dezember 2010; Veröffentlicht am: 18. Februar 2011 Dies ist eine Open-Access-Artikel unter den Bedingungen der Domain Erklärung Creative Commons Public verteilt, die besagt, dass, wenn in der Öffentlichkeit platziert, kann diese Arbeit frei reproduziert werden, verteilt, übertragen, verändert, als Grundlage oder auf andere Weise von jedermann zu jedem legalen Zweck verwendet Finanzierung:. Diese Studie wurde von der National Science Foundation (CCF-0621700, DBI0542119004, 1R01GM075331), Jilin Universität teilweise unterstützt wurde, die University of Georgia, die Georgia Cancer Coalition, die Georgia Research Alliance und die National Institutes of Health (1R01GM075331, DK69711). Die Geldgeber hatten keine Rolle in Studiendesign, Datenerfassung und Analyse, Entscheidung oder Vorbereitung des Manuskripts zur Veröffentlichung Konkurrierende Interessen:.. Die Autoren haben erklärt, dass keine Interessenkonflikte bestehen Einführung Die schnelle Weiterentwicklung von omic Vergleich zu Serum-Marker, vorhandenen Urinmarker sind meist konzentriert Harn-Trakt oder eng assoziierten Erkrankungen im Zusammenhang mit. Erst in den letzten Jahren verbessert hat proteomische Analyse von Urinproben ergab, dass, wie Seren, Urin ist auch eine reiche Informationsquelle für die Erkennung menschlicher Krankheiten wie dem Pfropf- gegen Marker Identifikation im Urin könnte möglicherweise durch vergleichende Proteomik-Analyse von Urinproben von Patienten mit einer bestimmten Krankheit und Kontrollgruppen durchgeführt werden. Die Herausforderung bei solchen sucht nach Urin-Marker in einer blind ist zweifach. (A) Urin konnte eine große Anzahl von Proteinen /Peptiden haben (im Gegensatz zum bisherigen Verständnis [8]) mit einer relativ geringen Menge. (B) Der Dynamikbereich in der Fülle dieser Proteine könnten einige Größenordnungen erstrecken, breiter ist als der Bereich, der typischerweise mit einem Massenspektrometer bedeckt [9]. Aus diesen Gründen vergleichende Analysen, insbesondere (semi) quantitative Analysen von Proteomik-Daten von Urinproben kann sehr schwierig sein. Dies könnte ein wichtiger Grund sein, dass es keine verlässlichen Urin-Marker für die Krebsdiagnose sind. Unsere Studie konzentriert sich auf die Entwicklung eines Berechnungsverfahren zur genauen Vorhersage der Proteine, die Urinausscheidungsorgane (siehe Abbildung 1 für den Umriss des Ansatzes sind ). Diese Proteine müssen bestimmte Eigenschaften aufweisen, die es ihnen ermöglichen, von den Zellen sekretiert wird und dann durch den Glomerulus Membran in Nieren ausgefiltert werden. Eine kürzlich Proteom Studie identifiziert mehr als 1.500 Proteine /Peptide, die in Urin durch gesunde glomerulären Membranen ausgeschieden werden [8]. Mit dieser Gruppe von Proteinen und Proteine als nicht Urin Ausscheidungsorgane zu sein, haben wir eine Liste der Unterscheidungsmerkmale zwischen diesen beiden Klassen von Proteinen identifiziert und ausgebildet, um eine Support-Vektor-Maschine (SVM), basierend Klassifikator zur Vorhersage, ob ein bestimmtes Protein könnte in den Urin ausgeschieden werden . Die Vorhersage-Methode wurde experimentell mit Western-Blots unter Verwendung von Antikörper-Arrays in Verbindung validiert, und die Ergebnisse sind sehr ermutigend. Diese Klassifizierer Proteine vorherzusagen angewendet wurde, die in den Urin ausgeschieden werden können, basierend auf den identifizierten differentiell exprimierten Gene in Magenkrebs im Vergleich zu Methoden Diese Studie besteht aus drei Hauptkomponenten:. (i) Aufbau eines Klassifikator für Urin Ausscheidungs Proteine vorherzusagen; (Ii) Bewertung der Leistung des Klassifikators, indem es einer Reihe von Proteinen, für die die Ausscheidungszustand der Proteine Anwendung bekannt ist; und (iii) Anwendung des validierten Klassifikator Gen-Expressionsdaten von Magenkrebs um seine Wirksamkeit zu zeigen, in der Urin-Marker Identifikationsproblem zu lösen. Diese Arbeit wurde von der Institutional Review Board an der University of Georgia genehmigt wurde, Athens, Georgia, USA (Amt des Vizepräsidenten für Forschung DHHS Assurance ID NO. FWA00003901, Projektnummer 2009-10705-1) und von der chinesischen Institutional Review Board der Überwachung am Menschen an der Jilin University College of Medicine, Changchun, China. Eine Einverständniserklärung, die von IRB an der University of Georgia und chinesischen IRB zugelassen, wurde von jeder Person erhoben. Alle Probanden sind sich bewusst, dass alle Daten aus der Forschung für Dokumente verwendet werden können, oder Publikationen wie in der Einwilligungserklärung angegeben. Das allgemeine Verständnis der Proteinausscheidung von Geweben Urin ist, dass einige Proteine oder sickerte aus den Zellen in den Blutkreislauf sezerniert werden, und dann ein Teil dieser Proteine, zusammen mit einigen nativen Proteinen in Blut kann in den Urin ausgeschieden werden. Unsere Ziele sind erste Unterscheidungsmerkmale für solche Urinausscheidungs Proteine zu identifizieren und dann einen Klassifikator zu bauen auf diesen Merkmalen beruhen, vorherzusagen, welche Proteine in Zellen können in den Urin ausgeschieden werden. Nach bestem Wissen und Gewissen, gab es weder im Falle einer Veröffentlichung richtet dieses Problem zu lösen. Die Bedeutung, die eine solche Fähigkeit ist, dass es eine wirksame Verbindung bei der Verbindung bietet omic der erste Schritt, eine solche Vorhersagefähigkeit bei der Entwicklung, dh ein Klassifikator, ist ein Trainingsdatenmenge enthält Proteine zu haben, und dass nicht in den Urin ausgeschieden werden können, auf deren Grundlage eine Reihe von Unterscheidungsmerkmale möglicherweise identifiziert werden konnten. Glücklicherweise haben wir einen großen Proteom-Datensatz von Urinproben von gesunden Menschen in einer kürzlich veröffentlichten Studie [8] gefunden, die mehr als 1.500 einzigartige Proteine enthält, von denen 1313 SwissProt Beitritt IDs. Wir haben diese 1313 Proteine als die positiven Trainingsdaten für die zu-trainierten Klassifikators verwendet. Das folgende Verfahren wurde dann ein negatives Trainingsset zum Generieren: willkürlich aus jeder Pfam Familie mindestens ein Protein auswählen, das keine positive Trainingsdaten enthält, und die Anzahl der ausgewählten Proteine aus jeder Familie ist auf die Größe der Familie proportional [ ,,,0],10], [11]. Als Ergebnis wurden 2.627 Proteine ausgewählt und als die negative Trainingssatz verwendet. Wir untersuchten 18 physikochemischen Eigenschaften von Proteinsequenzen berechnet, die potentiell nützlich für die Klassifikationsproblem sind auf dem allgemeinen Verständnis der Harnausscheidung von Proteinen beruhen . Die Details der 18 Merkmale und die Computerprogramme zu ihrer Berechnung sind in Tabelle S1 aufgeführt. Einige dieser Funktionen durch mehrere Merkmalswerte dargestellt werden, beispielsweise die Aminosäurezusammensetzung in einer Proteinsequenz wird von 20 Merkmalswerten dargestellt wird; Insgesamt sind die 18 Funktionen werden mit 243 Merkmalswerte dargestellt. Wir identifizierten dann eine Teilmenge von Merkmalen Werte aus der 243, die zwischen der positiven und der negativen Trainingsdaten unter Verwendung eines SVM-basierte Klassifizierer unterscheiden kann. Der RBF-Kernel wurde in unserem SVM Training verwendet wird, unter Berücksichtigung seiner Fähigkeit nichtlinearen Eigenschaften zu behandeln [12], [13]. Um welche der ursprünglich als Merkmale ermitteln tatsächlich nützlich sind, sofern das Feature-Auswahl-Werkzeug in LIBSVM [12] wurde verwendet, um die anspruchsvollsten Merkmale unter den 243. Weitere Funktionsauswahlwerkzeuge zur Auswahl möglicherweise verwendet werden könnte, aber wir haben viel Erfahrung in dieses Werkzeug verwenden und fanden es angemessen zu sein. Codes in dieser verwendet werden, sind öffentlich von LIBSVM Website (http://www.csie.ntu.edu.tw/~cjlin/libsvm/); wir haben auch das entsprechende Programm zugänglich http://seulgi.myweb.uga.edu/files gemacht. Ein F-Score [12], wie folgt definiert ist, wird verwendet, um die anspruchsvolle Leistung jedes Merkmalswert für unsere Klassifikationsproblem zu messen, Dabei bezieht sich auf die Trainingsmerkmalswerte (k = 1, ..., m); n Die Ausbildung unserer SVM-basierten Klassifikator erfolgt unter Verwendung eines Standardverfahrens zur Verfügung gestellt in LIBSVM [12] zu finden Werte von zwei Parametern C b. verwendet Datensätze, die Leistung des Klassifikators Eine unabhängige Datensatz wird die Leistung des trainierten Klassifizierer für die der Ausführungsstatus jedes Protein bekannt zu beurteilen wurde zur Bewertung verwendet. Die positive Teilmenge des Datensatzes hat 460 menschlichen Proteinen im Urin von gesunden Personen durch drei Urin Proteomik Studien [14], [15], [16] und die negative Untergruppe enthält 2.148 Proteine der gleichen Vorgehensweise wie zuvor ausgewählten verwenden, aber tut nicht mit dem negativen Satz für die Ausbildung verwendet überlappen die folgenden Maßnahmen verwendet wurden, um die Klassifikationsgenauigkeiten zu beurteilen. die Sensitivität, Spezifität, die Genauigkeit der Korrelationskoeffizient Matthew und die AUC [17]. Tabelle 1 fasst die Klassifikationsgenauigkeiten des trainierten Klassifizierer auf der sowohl die Ausbildung und die Testdatensätzen [17]. Von den Klassifikationsgenauigkeiten auf den beiden Datensätzen, glauben wir, dass unsere geschulten Klassifikator die wichtigsten unterschiedliche Merkmale der Ausscheidungs Proteine im Urin erfasst. Darüber hinaus wurde unser Klassifikator auf einem separaten Daten-Set getestet, eine Teilmenge der 274 Proteine auf einen vorgefertigten Protein-Antikörper-Array (die RayBio menschliche G-Serie Array 4000 (RayBiotech, Inc., Norcross, GA)) fixiert. Von den 274 Proteinen, 111 Ausscheidungsorgane zu sein, sind bekannt und wurden in unserer Ausbildung oder unabhängigen Testdatensätzen enthalten. Wir legten den Klassifikator auf den verbleibenden 163 Proteine, für die die Ausscheidungsorgane Status unbekannt war (siehe Ergebnisse und Tabelle S2). Diese Protein-Array stellt die relativen Expressionsniveaus für jedes Protein auf der Anordnung, wenn sie auf einem (Urin) zu testende Probe, die durch die Densitometrie in Bezug auf die Signalintensität quantifiziert wird gemessen. Der Hintergrund des Arrays wurde als Kontrolle verwendet, um die tatsächliche Anwesenheit eines Proteins in die (Harn) Probe zu bestimmen. Die Signalintensität für ein Protein wurde als ein wahres Signal angesehen, wenn sie mindestens 5-fach höher war als die der Kontrolle, wie von den Empfehlungen des Herstellers vorgeschlagen. Wir konzentrierten uns unsere experimentellen Validierung auf der positiven Voraussagen bestätigen nur, da es praktisch unmöglich ist, ein Protein zu beweisen, nicht aufgrund von Einschränkungen in der Detektionsempfindlichkeit der gegenwärtigen Technologie in einer Urinprobe vorhanden ist, wenn das Protein von sehr niedriger Konzentration in der Probe ist. c. Urinprobenentnahme /der Zubereitung Urinproben von Magenkrebs-Patienten und gesunden Kontrollen wurden an der Medizinischen Fakultät der Universität Jilin, Changchun, China gesammelt. Magenkrebs-Patienten, von wem die Proben aus gesammelt wurden, sind alle späten Stadium Patienten (siehe Tabelle S3 für Patienteninformationen). Diese Proben wurden sofort lyophilisiert und bei -80 ° C bis zur weiteren Verwendung gelagert und nach der chirurgischen Entfernung von den Patienten. Sie wurden dann rekonstituiert und zentrifugiert (3.000 xg Insgesamt 80 Magenkrebsgewebe und ihren benachbarten noncancerous Gewebe von 80 Patienten an der Medizinischen Fakultät der Universität Jilin wurden gesammelt. Microarray-Experimente wurden an diesen Geweben durchgeführt, die Affymetrix Genechip Menschen Exon 1.0 ST Array mit, die 17.800 menschlichen Gene abdeckt. Der PLIER Algorithmus [18] wurde verwendet, um die Sondensignale auf Gen-Ebene Ausdrücke zusammenzufassen. Für jedes Gen untersuchten wir die Verteilung der Expression fold change zwischen den paarigen Krebs und Kontrollgeweben über alle 80 Paare von Geweben. Lassen Sie K exp, Die DAVID Bioinformatik-Ressourcen und die KOBAS Web-Server [20], [21] wurden zu tun, funktionale und Pathway-Anreicherung Analyse verwendet wurden, für alle vorhergesagten Urin-Ausscheidungs-Proteine, die unter Verwendung von ganze Reihe von menschlichen Proteinen als Hintergrund. Wir verweisen den Leser auf [20], [21] Einzelheiten zu den Methoden für die funktionale und Pathway-Anreicherung analysiert. Mit DAVID Bioinformatik-Ressourcen, die Anreicherung Punktzahl für eine bestimmte Gruppe von Proteinen wurde durch die EASE-Score bestimmt [20], [22]. KOBAS ist ein ergänzendes Instrument zu DAVID, da sie die Gen-Annotation mit KEGG Orthologie (KO) Begriffe erweitert. Der KOBAS Web-Server, zusammen mit dem KO-basierten Kommentarsystem [21], [23], wurde verwendet, um statistisch angereichert und unterrepräsentierte Wege unter den vorhergesagten Urin ausgeschiedenen Proteine finden. KOBAS nimmt in einer Reihe von Proteinsequenzen und annotiert sie die KO Begriffe verwenden. Die kommentierten KO Begriffe wurden dann verglichen gegen alle menschliche Proteine als Hintergrund-Set für die Beurteilung, ob sie angereichert sind oder unterrepräsentiert. Harnproteine von jeder Probe (insgesamt 2 ug) wurden mit 3x Probe Farbstoff kombiniert. Jedes Röhrchen wurde 5 min gekocht und geladen auf SDS-PAGE-Gelen, zusammen mit 10 ul Standards und für 1 h bei 200 Volt laufen. Die Membran wurde mit 100% Methanol aktiviert, nach einer Übertragung von dem Gel auf die Membran (100 Volt für 1 h). Sobald die Übertragung abgeschlossen war, wurde die Membran zu trocknen, wieder angefeuchtet in 100% Methanol gewaschen und erlaubt 2X für jeweils 5 min mit Tris-gepufferter Kochsalzlösung (TBS). Die Membran wurde dann 2 h bei Raumtemperatur in 3% Milchblockierungslösung inkubiert. Als nächstes wird die Membran in der ersten Antikörper-Lösung (1:200 Verdünnungen in 1,5% Milch-blocking) für 1 h bei Raumtemperatur, und die ungebundenen Antikörper inkubiert wurde 10 durch Waschen der Membran 3 x mit TBS Tween-20 (TBST) Lösung entfernt min je. Dann wurde die Membran in einer 1:10,000 Verdünnung des sekundären Antikörpers in 1,5% Milch Blockierungslösung für 1 h bei Raumtemperatur inkubiert. Die Membran wurde 3 x mit TBST und 2X mit TBS (jeweils 10 Minuten) gewaschen. Schließlich wurde die Membran vollständig mit einer gleichen Menge von Enhancer und Peroxid-Lösung aus einem Pierce Western Blotting Kit für 5 min und belichtet den Film bedeckt. Jedes Experiment wurde mehrmals wiederholt, um die Reproduzierbarkeit [24] gewährleisten. Die Signalintensitäten wurden mit dem ImageJ Software [25] bestimmt. Für jede Membran wurde die leere Spur verwendet, um die Signalintensitäten über die Membranen zu normalisieren. Die Performance wurde unter Verwendung von ROC und Whisker-Box-Plot untersucht. a. Signalpeptid und Sekundärstrukturen sind wesentliche Merkmale von Urin ausgeschiedenen Proteine Die erste Liste von Funktionen sorgfältig ausgewählt wurde, was Ihnen an Proteineigenschaften relevant Urin ausgeschieden werden angenommen, basierend auf Literatursuche und unser gegenwärtiges Verständnis von Harn- Proteinen. Zum Beispiel wird die negativ geladene Wand glomerulären Nieren in der Filtration von erlauben nur positiv oder neutral geladene Proteine. Somit ist verantwortlich für ein Protein eines der Features, die wir ausgewählt. Unter den verfügbaren Informationen in Betracht zieht, war die Gesamtzahl der Merkmalswerte gesammelt anfänglich 243, was Grundsequenzeigenschaften, Motive, physikalisch-chemischen Eigenschaften und strukturellen Eigenschaften (Tabelle S1). Bei der Identifizierung von Merkmalen, die Urin wirksam sind Ausscheidungs-Proteine von den nicht-Ausscheidungs diejenigen, eine einfache und effektive Methode, um Funktionen zu beseitigen, die zeigen wenig oder keine anspruchsvolle Leistung für unser Klassifikationsproblem beschäftigt war bei der Unterscheidung; 74 Merkmalswerte wurden unter Verwendung des gemäß der Beschreibung in Abschnitt A-Methoden (Tabelle S5) ausgewählt. Diese Merkmalswerte wurden verwendet, um die endgültige Klassifikator trainieren. Unter den ausgewählten Merkmale, die diskriminierend war die Anwesenheit von Signalpeptiden. Es versteht sich, dass Proteine, die Signalpeptide durch das ER und sind befahrene zu ihrem Bestimmungsort entsprechend den spezifischen Signalpeptide sezerniert werden; also nicht überraschend, dass die meisten ausgeschiedenen Proteine haben diese Funktion. Ein weiteres herausragendes Merkmal war die Sekundärstruktur-Typ; Insbesondere wurde als die Zahl 2 Merkmalswert unter den ausgewählten 74 (Tabelle S5) in der Reihenfolge der Anteil der alpha-Helices in einer Proteinsequenz. Wie erwartet, war die Ladung eines Proteins unter den bestplatzierten Funktionen für ausgeschiedene Proteine. Dies ist konsistent mit dem allgemeinen Verständnis, dass Ladung ist ein Faktor bei der Bestimmung, welche Proteine können durch die glomerulären Membran filtriert werden [26] als Proteine innerhalb der glomerulären Membranen und podocyte Schlitze sind negativ geladen, und somit negativ geladenen Proteine geringe Chancen haben, zu filtern, durch die Nieren. Tatsächlich waren die Merkmalswerte der positiven Aminosäuren und Ladung unter den bestplatzierten Merkmalswerte. Interessanterweise jedoch Molekulargewicht, die bei 232 von 243 rangiert, wurde nicht in den letzten 74 Merkmalswerte enthalten. Dies könnte durch die nachfolgend erläutert werden. Die Proteine im Serum kann bereits eine Spaltung durchlaufen oder wurden teilweise abgebaut und somit nicht in ihrer intakten oder vollständigen Form sein kann, wenn sie die Niere ein. Es wurde in der Tat festgestellt, dass die Mehrheit der Proteine im Urin gefunden extensiv abgebaut werden [27]. Während ein intaktes Protein nicht in der Lage sein kann, wegen seiner Größe oder Form, einem Protein abgeleitetes Peptid durch den Glomerulus zu filtern können leicht durch die podocyte Schlitze passieren. Als Ergebnis ist das Molekulargewicht des intakten Proteins eine nicht-Faktor bei der Vorhersage, wenn das Protein Urin Ausscheidungsorgane ist. Es ist zu beachten, dass Urin Ausscheidungs Proteine und sekretierte Proteine einige gemeinsame Merkmale, wie einige der Aktie verwendeten Funktionen blood-sekretierten Proteine in unserer früheren Studie [10] wurden ausgewählt, in der Urin-Protein-Prädiktion in dieser Untersuchung zu identifizieren. Zum Beispiel Funktionen wie Lösungsmittel Zugänglichkeit, Polarität und Signalpeptide wurden in beiden Klassifizierer enthalten. Allerdings gibt es einen deutlichen Unterschied zwischen den in den beiden Klassifizierer verwendeten Funktionen. Während Features wie Beta-Strang-Inhalt bietet mit Beta-Barrel-Transmembranprotein und Protein-Verhältnis verbunden sind, TATP Motiv, Transmembran-Domäne, Proteingröße und die längste ungeordneten Bereich unter den Top-Features waren für die Vorhersage von Blut-sekretorischen Proteinen [10 ], wurden sie in den letzten Features für die Urin-Protein-Prognose nicht enthalten. Darüber hinaus auf positive Ladung bezogene Funktionen, wie die Zusammensetzung von positiv geladenen Aminosäuren, waren prominent in Urin-Protein-Vorhersage, aber nicht im Blut Sekretion Vorhersage ausgewählt. Ebenso wurden die alpha-Helix-Gehalt und die Spule-Gehalt an Proteinen unter den Top-Features für Urin-Protein-Vorhersage, aber sie waren nicht für das Blut-sekretorischen Protein Vorhersage ausgewählt. Interessant ist, dass im Gegensatz zu der Feststellung zu beachten, dass beta-Stränge eine gemeinsame Sekundärstrukturtyp unter den Blut sekretorische Proteine sind, Harn- Proteine neigen höhere alpha-Helix und Spule Gehalt zu haben, die anzeigt, dass die Urin-Proteine besitzen Eigenschaften nicht geteilt von Blut sekretorischen Proteinen im allgemeinen. b. Die Leistung des Klassifikator Um die Genauigkeit des endgültigen Klassifizierer bestimmen, die wir getestet es auf einem unabhängigen Test-Set, das experimentell validiert Urin Ausscheidungs Proteine von 460 besteht und 2148 nicht Urin Ausscheidungs Proteine. Unsere Klassifikator hat seine Prognose Sensitivität und Spezifität auf dieser unabhängigen Testsatz bei 0,78 und 0,92, bzw. (Tabelle 1). Wir liefen dann den Klassifikator auf der 163 von den 274 Proteinen auf der vorgefertigten Antikörper fixiert Array (siehe Methoden), für die der Ausführungsstatus unbekannt war. Von den 163 Proteinen wurden 112 Proteine vorhergesagt Urin Ausscheidungsorgane von unseren Klassifikator zu sein. Zur Beurteilung der Leistung dieser Vorhersage Antikörper Array-basierten Experimenten an 14 Urinproben durchgeführt, sieben von gesunden Personen und sieben von Magenkrebs-Patienten. Von den 112 vorhergesagt Urin-Ausscheidungs Proteine, 92 wurden in mindestens einem der Urinproben (Tabelle S6), was eine positive Vorhersagerate von 0,81, gefunden, die mit dem Leistungsniveau auf dem ersten Test-Set konsistent ist. es ist zu beachten, dass eine Einschränkung dieses Klassierers ist, dass einige Proteine könnten teilweise abgebaut werden, bevor mit dem Urin ausgeschieden oder im Urin, was es schwierig macht für unsere Klassifikator zu erfassen, so gebildeten Peptide, wie sie auf ganze intakte Proteine ausgebildet wurde. Dieses Problem wird in Zukunft durch Ableitung Merkmalswerte auf der Grundlage der tatsächlichen Proteine /Peptide in früheren Urin-Proteom-Studien identifiziert angesprochen werden, anstatt ihre entsprechenden Proteine in voller Länge, wie in dieser Studie durchgeführt. Zwar gibt es deutlich Raum für weitere Verbesserungen ist, sind die Vorhersageergebnisse des aktuellen Klassifikator sehr ermutigend. Unsere früheren Studie über 160 Arten von Microarray-Gen-Expressionsdaten von Magenkrebs hat 715 differentiell exprimierten Genen, die mit mindestens 2-fache Veränderungen im Magen-Krebs identifiziert im Vergleich zu Für (i) haben wir durchgeführt funktionelle und Wege Anreicherung Analysen auf allen 201 Proteine, die die DAVID mit [20 ] und KOBAS [21] Server, respectively. Wir fanden, dass die angereicherten funktionellen Gruppen der extrazellulären Matrix enthalten (ECM), die Zelladhäsion und Entwicklung, Zellmotilität, Abwehrreaktion, Angiogenese, die alle in der Entwicklung oder in der Abwehr von Krebserkrankungen (Abbildung S1A) beteiligt zu sein, sind bekannt. Die angereicherten Wege waren ECM-Rezeptor-Interaktion und anorganischen Ionentransport und Stoffwechselwege (Abbildung S1B) Das folgende Kriterium verwendet wurde, um die Liste der 201 Proteine, die für die Schritte (ii) zu reduzieren - (iii). haben die Proteine nicht berichtet worden, einem Krebs auf der Basis unserer umfangreichen Literaturrecherche Wir wählten sechs Proteine (MUC13, Col10a1, AZGP1, LIPF, MMP3 und EL) für experimentelle Validierung der oben verengt Liste. Um dies zu tun, haben wir Urinproben von 21 Patienten mit Magenkrebs und 21 gesunden Personen gesammelt. Von den sechs ausgewählten Proteine, fünf Proteine, MUC13, Col10a1, LIPG, AZGP1 und EL wurden durch Western-Blots in mindestens einer Urinprobe detektiert. Von den fünf, MUC13, Col10a1 und EL wurden selbst bei einer sehr geringen Menge der Gesamturinproteine (1-2 &mgr; g) nachgewiesen. MMP3 wurde nicht in den Proben fanden wir getestet, die im Urin oder einer falschen Vorhersage von unseren Klassifikator der niedrigen Konzentration von MMP3 zurückzuführen sein kann. Es ist besonders interessant, dass wir in der Lage waren konsistent Unterschiede zu erkennen in der EL Überfluß (codiert durch LIPG Das Molekulargewicht dieses Proteins bestimmt wurde, 68 kDa zu sein [28]. Somit ist ein Homo-Dimer erwarteten 134 kDa zu sein. In den Western-Blots wurden jedoch Banden bei nahezu 100 kDa nachgewiesen. Dies entspricht wahrscheinlich einer teilweise gespalten homo-Dimer, eine aktive Form, die von einer früheren Studie bestätigt wurde [29], obwohl die Möglichkeit einer monomeren Form von EL mit einem anderen Protein verbunden sind, können nicht ausgeschlossen werden. http://csbl.bmb.uga.edu/~juancui/Publications/GC2009/Additional_material.pdf.
Kontrollgewebe kann dieses Verfahren verwendet werden, für die Krankheit Potential Urin Marker vorherzusagen. Hier berichten wir über die detaillierten Algorithmus dieser Verfahren und eine Anwendung zur Identifizierung von Urin Marker für Magenkrebs. Die Leistung des trainierten Klassifizierer auf 163 Proteine wurde unter Verwendung von Antikörper-Arrays experimentell validiert, zu erreichen > 80% wahre Positive-Rate. Durch Anwendung des Klassifikators auf differentiell exprimierte Gene in Magenkrebs vs
normalen Magengewebe, wurde gefunden, dass die endotheliale Lipase (EL) wurde in den Urinproben von 21 Patienten mit Magenkrebs wesentlichen unterdrückt gegen
21 gesunden Personen. Insgesamt haben wir gezeigt, dass unsere Prädiktor für Urin Ausscheidungs Proteine ist sehr effektiv und könnte möglicherweise als ein mächtiges Werkzeug bei der Suche nach Krankheit Biomarker in Urin im Allgemeinen dienen
Techniken in den letzten Jahren hat es möglich gemacht, Biomarker für bestimmte Krankheiten des Menschen auf systematische und umfassende Weise zu suchen, die im wesentlichen unsere Fähigkeit verbessert zu erkennen Krankheiten bei frühe Stufen. Die meisten der früheren Studien haben sich auf Biomarker Serummarker [1], vor allem wegen der bekannten Vielfalt von Serum in haltigen Signale für verschiedene physiologischen und pathophysiologischen Bedingungen.
-host Krankheit und koronare Herzkrankheit [2], [3], [4]. Man beachte, dass Urin durch Filtration von Blut durch die Nieren gebildet wird; daher einige Proteine im Blut kann durch die Filter passieren und in den Urin ausgeschieden werden. Als Ergebnis spiegeln die Urinproteine nicht nur die Bedingungen der Niere und des Urogenitaltraktes, sondern auch die der anderen Organe, die von der Niere distal sein kann, wie mindestens 30% der Urinproteine nicht ursprünglich aus dem Urogenitaltrakt sind [5], [6]. Die Fülle von Informationen im Urin eine attraktive Quelle für Biomarker-Screening macht, da im Vergleich zu Serum ist die Zusammensetzung des Urins relativ einfach und Urinsammlung ist einfacher und nicht-invasive [7], [8].
Referenzmagengewebe; und eine Anzahl von potentiellen Urinmarker für Magenkrebs wurden identifiziert. Ein wesentlicher Beitrag dieser Arbeit gemacht ist, dass es eine neue und effektive Möglichkeit bietet, indem darauf hindeutet Kandidaten Markerproteine Proteom Studien von Urin zu führen, so die Möglichkeit, gezielt Marker sucht Antikörper-vermittelte Techniken wie Western-Blots und Elisa unter Verwendung, die wesentlich mehr machbar sind als groß angelegte vergleichende Proteomik Analysen von Urinproben ohne Ziele, mit denen zu arbeiten. Während dieses Vorhersageprogramm zu Magenkrebs Daten in dieser Studie keine Magenkrebs-spezifische Informationen in diesem Programm verwendet angewandt worden sind; daher kann es auch für andere Krankheiten, für Urin-Marker sucht verwendet werden
a. Ein Algorithmus für die Ausscheidungs-Proteine die Vorhersage
von Geweben zu Markierungssuche in Urin-Analysen von Kandidatenmarker im Urin die Bereitstellung, die unter Verwendung von Antikörper-basierten Ansätzen untersucht werden können.
+ und n
- sind die Anzahl der Proteine, die in der positiven (+) und negativen (-) Trainingsdaten sind; , Sind die Mittelwerte der i
ten Merkmalswert in der gesamten Trainingsdatenmenge, die positive Datensatzes und der negativen Datensatzes sind; und und sind die i
th Funktion des k
th Protein in den positiven und negativen Trainingsdaten sind. Im Allgemeinen, desto größer ist ein F-Score, desto mehr diskriminativen die entsprechende Funktion. In unserer Auswahl werden alle Funktionen mit F-Werte über einer vorgewählten Schwelle wurden in die Ausbildung der Endklassifizierers erhalten und verwendet. Um eine optimale F-Score Schwelle zu finden, als wir eine Liste der möglichen Schwellenwerte und dann ausgewählt, um die beste auf der Grundlage der Trainingsergebnisse.
und γ, die eine optimale Einteilung auf den Trainingsdaten geben, wobei C
den Trade-off zwischen steuert Trainingsfehler und Klassifizierung Margen und γ bestimmt die Breite des Kernels verwendet [12]. Unser Trainingsprozedur wird wie folgt zusammengefasst [12]:
und γ, und dann gilt es an die Untervalidierungsdaten und die Berechnung der Klassifikationsfehler zu suchen;
für 25 min bei 4 ° C) Zellkomponenten zu entfernen. Die Überstände wurden gesammelt und bei 4 ° C gegen Millipore Reinstwasser (drei Pufferwechsel durch eine Dialyse über Nacht gefolgt) mit Slide-A-Lyzer Dialyse Cassetten (Thermo Fisher Scientific, Rockford, IL) dialysiert. Proteinkonzentrationen wurden mittels des Bio-Rad Protein Assay (Bio-Rad, Hercules, CA) unter Verwendung von mit Rinderserumalbumin als Standard.
d. Identifizierung von Genen, die differentiell exprimiert bei Magenkrebs und Kontrollgewebe sind
die Anzahl der Paare von Geweben sein, deren fold change mindestens 2. Ein Gen wird als unterschiedlich exprimiert
, wenn die p
-Wertes des beobachteten K exp
weniger als 0,05. Mit diesem Kriterium wurden insgesamt 715 Gene gefunden bei Magenkrebs in allen menschlichen Gene unterschiedlich exprimiert werden, und die Namen der 715 Gene, zusammen mit der zugehörigen K exp
und p
-Werte, sind in Tabelle S4 gegeben. Eine detaillierte Studie des Microarray-Daten wurde an anderer Stelle berichtet [19].
e. Funktion und Pathway-Anreicherung analysiert
f. Western-Blots
Ergebnisse und Diskussion
c. Die Anwendung der Klassifikator zu Magenkrebs Daten
Kontrollgewebeproben [19]. Während es vorzuziehen wäre, Proteom-Daten der Gewebeproben zu haben, haben wir nur Gen-Expressionsdaten in dieser Studie zur Verfügung. Daher werden Daten, die Genexpression als Annäherung an die Protein-Expression in dieser Methodik orientierten Studie verwendet. Unserem Klassifizierer wurde auf diese 715-Proteine angewendet und vorhergesagt, daß 201 der 715-Proteine sind Urinausscheidungsorgane. Tabelle S7 stellt die detaillierte Informationen über die 201-Proteine. Da es unrealistisch ist, alle 201 Proteine, die in dieser Studie zu überprüfen, um zu bestimmen, ob sie Urin Ausscheidungsorgane sind oder nicht, analysiert wir haben diese Liste zu verengen. Im Einzelnen haben wir folgende Analysen durchgeführt: (i) funktionelle und Pathway-Anreicherung analysiert ein besseres Verständnis der verschiedenen Arten von Proteinen im Urin zu gewinnen, (ii) Literatursuche zu Harnproteine Informationen über veröffentlichte Urinmarkerproteine zu kompilieren, ( iii) die Genexpressionsdaten untersuchen Gene zu entfernen, die nicht wesentlich unterschiedlich zwischen Krebs und Kontrollgewebeproben ausgedrückt; und (iv) Western Blots auf Proteine aus einer verengten Down-Liste der 201 Proteine ausgewählt. Dieses Verfahren zeigte eine hohe Erfolgsrate und führte zu einer interessanten Entdeckung potentieller Biomarker für Magenkrebs.
bezogen werden, was zu 71 Proteine gibt. Die Liste wurde weiter auf der Basis eines vorgewählten Cutoff auf Differentialausdrücke und funktionellen Annotationen (potentiell relevant für Magenkrebs und nicht-Immunantworten) reduziert.
d. Endothelial Lipase wird in den Urinproben von Patienten mit Magenkrebs
erheblich reduziert
) zwischen den beiden Sätzen von 21 Urinproben. Die Western-Blots für EL zeigte eine deutliche Reduktion in seiner Fülle in den Urinproben der 21 Magenkrebs-Patienten im Vergleich zu den Kontrollproben. Wie in 2A gezeigt ist, zeigte die Mehrheit der Kontrollproben von EL die Anwesenheit, während die meisten der Magenkrebsproben relativ geringe Mengen an EL hatte. Dieses Muster wurde wiederholt beobachtet
doi:10.1371/journal.pone.0016875.s005
(XLS)
Table
 Anti-Coronavirus-Moleküle von Mikroben könnten der Schlüssel zu neuen Behandlungen sein
Anti-Coronavirus-Moleküle von Mikroben könnten der Schlüssel zu neuen Behandlungen sein
 Infliximab kann die Wirksamkeit einiger COVID-19-Impfstoffe beeinträchtigen
Infliximab kann die Wirksamkeit einiger COVID-19-Impfstoffe beeinträchtigen
 Apoptose ist ein wichtiger Mediator der Pathogenese bei der Infektion mit dem Coronavirus bei Tieren
Apoptose ist ein wichtiger Mediator der Pathogenese bei der Infektion mit dem Coronavirus bei Tieren
 Warum brauche ich eine Koloskopie?
Warum brauche ich eine Koloskopie?
 Was ist der Deal mit Hepatitis C?
Was ist der Deal mit Hepatitis C?
 Ländliche und städtische Mikrobiota unterscheiden sich schon in jungen Jahren,
Ländliche und städtische Mikrobiota unterscheiden sich schon in jungen Jahren,
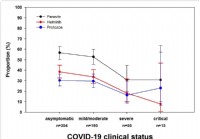 Untersuchungen zeigen, dass ein Befall mit Darmparasiten den Schweregrad von COVID-19 verringert
Wir lernen jeden Tag mehr über die COVID-19-Krankheit. Erwachsene jeden Alters mit bestimmten Grunderkrankungen haben ein erhöhtes Risiko für schwere Erkrankungen durch das Virus, das COVID-19 verursa
Untersuchungen zeigen, dass ein Befall mit Darmparasiten den Schweregrad von COVID-19 verringert
Wir lernen jeden Tag mehr über die COVID-19-Krankheit. Erwachsene jeden Alters mit bestimmten Grunderkrankungen haben ein erhöhtes Risiko für schwere Erkrankungen durch das Virus, das COVID-19 verursa
 Pflanzliche Lebensmittel können antibiotikaresistente Superbakterien auf den Menschen übertragen
Forscher der University of Southern California (USC) haben auf der Jahrestagung der American Society for Microbiology gezeigt, dass pflanzliche Lebensmittel Antibiotikaresistenzen auf das Darmmikrobio
Pflanzliche Lebensmittel können antibiotikaresistente Superbakterien auf den Menschen übertragen
Forscher der University of Southern California (USC) haben auf der Jahrestagung der American Society for Microbiology gezeigt, dass pflanzliche Lebensmittel Antibiotikaresistenzen auf das Darmmikrobio
 Immunzellen reparieren geschädigten Darm bei Kindern mit CED
Laut einer neuen Studie, die in der Zeitschrift veröffentlicht wurde Gastroenterologie , Immunzellen einer bestimmten Art, die Entzündungsprozesse regulieren und sogar dazu beitragen, die normale Da
Immunzellen reparieren geschädigten Darm bei Kindern mit CED
Laut einer neuen Studie, die in der Zeitschrift veröffentlicht wurde Gastroenterologie , Immunzellen einer bestimmten Art, die Entzündungsprozesse regulieren und sogar dazu beitragen, die normale Da