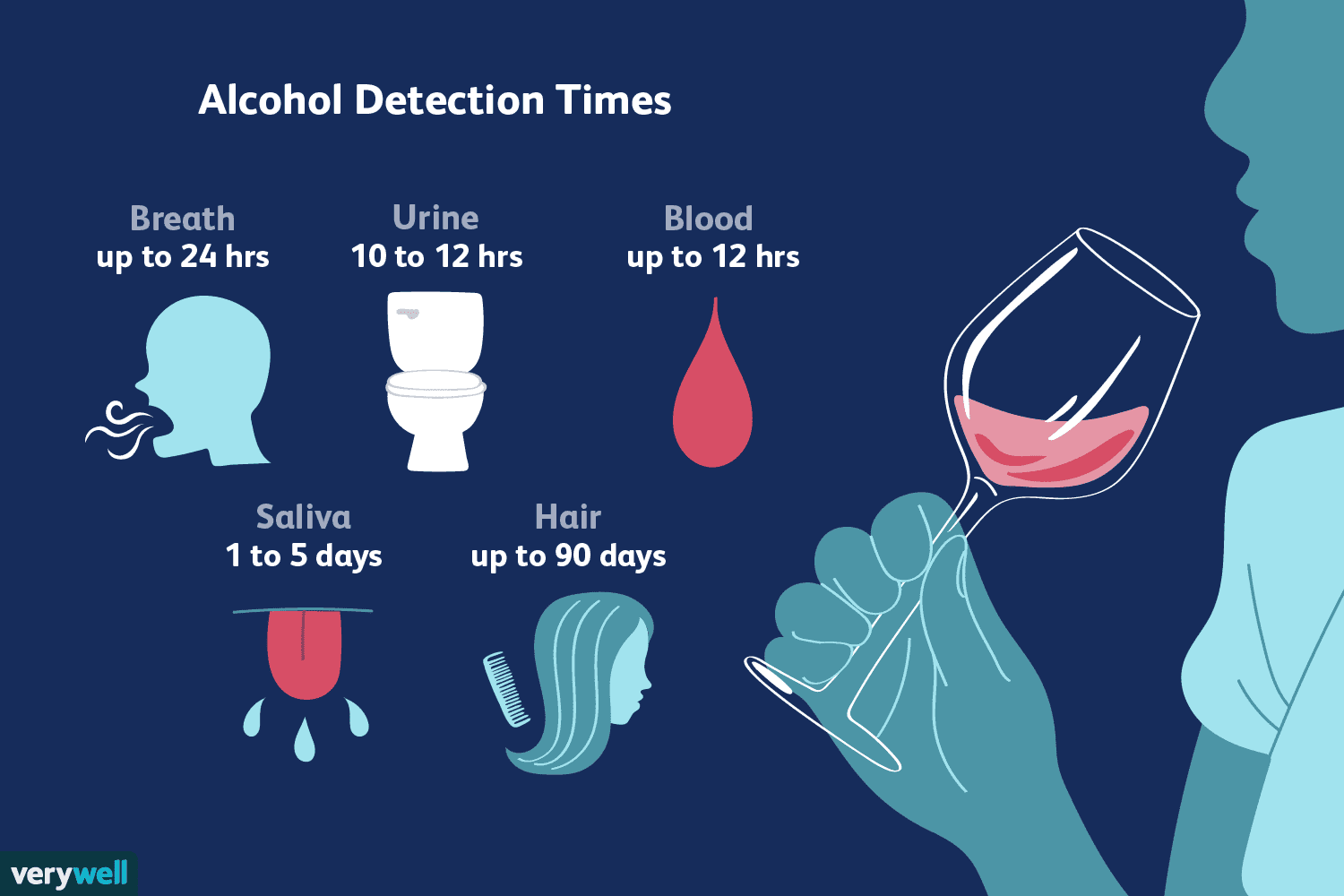
personalizzati sequenziamento profondo del carcinoma gastrico rivela mutazioni somatiche rilevanti per la medicina personalizzata
Abstract
sfondo
A livello globale, il cancro gastrico è la seconda causa più comune di morte per cancro , con la maggior parte degli oneri a carico della salute dei paesi economicamente meno sviluppati
Metodi
Qui., riportiamo una caratterizzazione genetica di 50 campioni con adenocarcinoma gastrico, utilizzando gli array Affymetrix SNP e array di espressione di mRNA Illumina così come Illumina sequenziamento delle regioni codificanti di 384 geni appartenenti a vari percorsi noti per essere alterato in altri tipi di tumore.
Risultati
alterazioni genetiche sono state osservate in Wnt, Hedgehog, ciclo cellulare, danno al DNA e epiteliali-to-mesenchimale transizione percorsi
. Conclusioni
i dati suggeriscono terapie approvate mirati o in fase di sviluppo clinico per carcinoma gastrico sarebbe di beneficio per ~ 22% dei pazienti studiati. Inoltre, le nuove mutazioni rilevate qui, sono suscettibili di influenzare la risposta clinica e suggerire nuovi bersagli per la scoperta di nuovi farmaci.
Sfondo
Nonostante la recente diminuzione dei tassi di mortalità da cancro gastrico in Nord America e nella maggior parte settentrionale e occidentale , cancro allo stomaco rimane una delle principali cause di morte in tutto il mondo ed è comune in Giappone, Corea, Cile, Costa Rica, Federazione russa e in altri paesi dell'ex Unione Sovietica [1]. Nonostante i miglioramenti nella modalità di trattamento e di screening, la prognosi dei pazienti con adenocarcinoma gastrico rimane poveri [2]. Per comprendere la patogenesi e di sviluppare nuove strategie terapeutiche, è essenziale per sezionare i meccanismi molecolari che regolano la progressione del cancro gastrico. In particolare, i meccanismi oncogenici, che può essere bersaglio di medicina personalizzata.
Il termine "dipendenza oncogene" per descrivere le cellule tumorali altamente dipendenti da un dato oncogene o via oncogenica è stato introdotto da Weinstein [3, 4]. Il concetto sottolinea lo sviluppo di terapie mirate che tentano di inattivare un oncogene, fondamentale per la sopravvivenza delle cellule tumorali, mentre risparmiando le cellule normali che non sono allo stesso modo assuefatti.
Diversi oncogeni attivati ad alta frequenza in altri tipi di tumore sono stati anche dimostrato di essere mutato nel cancro gastrico. Ne consegue che la terapeutica commercializzati destinate a questi oncogeni avrebbero trattare efficacemente una percentuale di carcinomi gastrici, sia come agenti singoli o in combinazione. Nel gennaio 2010, trastuzumab è stato approvato in combinazione con la chemioterapia per il trattamento di prima linea del carcinoma gastrico ERBB2
-positivo avanzato e metastatico. Trastuzumab è il primo agente mirato ad essere approvato per il trattamento del carcinoma gastrico ed un incremento del 12,8% nel tasso di risposta è stato visto con l'aggiunta di trastuzumab alla chemioterapia in ERBB2
adenocarcinoma gastrico positivo [5, 6]. E 'stato stimato che il 2-27% dei tumori gastrici porto ErbB2
amplificazioni e può essere trattata con inibitori ErbB2 [7, 8]. Allo stesso modo, la sovraespressione di un altro recettore tirosin-chinasi (RTK) EGFR
, è stato notato nel cancro gastrico e le prove multiple di EGFR
inibitori di questo tipo di cancro sono in corso (rivisto in [9, 10]). Inoltre alcuni tipi di cancro gastrico porto amplificazione del DNA o sovraespressione della RTK MET
[11, 12] e la sua paralogo MST1R
[13] e possono essere trattati con il TEM
o MST1R
inibitori [14-20 ]. Infine, FGFR2
sopra espressione e l'amplificazione è stata osservata in una piccola percentuale di tumori gastrici (scirrhous) [21] e gli inibitori hanno dimostrato una certa efficacia in clinica [22].
A valle delle RTK, KRAS
l'amplificazione di tipo selvatico e la mutazione è stata trovata anche in circa il 9-15% dei tumori gastrici [23, 24] e possono essere efficacemente trattati con inibitori MEK [25, 26]. L'attivazione della via PI3K /AKT /mTOR è stato anche visto in 4-16% del cancro gastrico [27-30] e così può essere sensibile agli inibitori della PI3K [31-34]. Allo stesso modo, ciclo cellulare chinasi AURKA
ha dimostrato di essere attivato nel carcinoma gastrico [35, 36] e gli inibitori AURKA in sviluppo clinico [37] possono avere un beneficio clinico.
Rapporti della frequenza di diversi tipi di attivazione oncogenica e loro co-occorrenza sono limitati. In contrasto con gastrointestinonal tumori stromali (GIST) che sono caratterizzati da un'alta frequenza di KIT
e PDGFRA
attivazione [38] e, quindi, trattati efficacemente nella maggior parte da imitanib e sunitinib [39, 40], appare adenocarcinoma gastrico una malattia eterogenea molecolarmente senza alta frequenza perturbazione oncogeno scoperto finora. Questo è illustrato da una recente indagine di mutazione somatica nei geni codificanti chinasi in 14 linee di cellule di cancro gastrico e tre i tessuti di cancro gastrico che hanno rilevato variazioni di singoli nucleotidi più di 300 nuovi chinasi e varianti strutturali chinasi-related. Tuttavia, la mutazione non molto frequentemente ricorrenti o chinasi mutato è stata scoperta [41].
Con l'obiettivo di chiarire il potenziale per il trattamento di carcinoma gastrico con terapie mirate sia sul mercato, in fase di sviluppo o da scoprire, abbiamo caratterizzato clinica campioni di carcinoma gastrico per rilevare attivazione di oncogeni.
Abbiamo preso un approccio globale analizzando i campioni in Affymetrix array SNP e array di espressione di mRNA Illumina. Queste tecnologie sono ben validati per la rilevazione del genotipo, DNA variazione del numero di copie e mRNA profilo di espressione. Esse sono suscettibili di campioni clinici eterogenei. I campioni sono stati interrogati anche da (Illumina) sequenziamento di seconda generazione. Relativamente nuove tecnologie di sequenziamento di seconda generazione offrono sia maggiore produttività e la capacità di sequenziamento profondo. Quest'ultimo è particolarmente importante per caratterizzare campioni tumorali che tendono a includere una miscela di tipi cellulari compresi infiltranti cellule normali, e vascolare delle cellule tumorali di diversi genotipi. In questo studio abbiamo utilizzato l'arricchimento di destinazione e la tecnologia di sequenziamento Illumina per sequenziare le regioni codificanti di 384 geni. Abbiamo deciso di privilegiare la profondità di copertura su più ampia copertura al fine di cogliere le mutazioni presenti in sottopopolazioni all'interno dei tumori. Recenti studi hanno dimostrato tumori tendono a nutrire molte mutazioni in un numero minore di vie di segnalazione [42, 43] quindi ci siamo concentrati sui geni in queste vie. Abbiamo anche incluso geni che codificano per le proteine precedentemente dimostrato di influenzare la risposta alle terapie mirate e più probabilità di essere presi di mira con successo da piccolo intervento molecola, come il nostro obiettivo è quello di trovare modi più efficaci e innovativi di trattamento del carcinoma gastrico.
Metodi
Tissue campioni
campioni di DNA e RNA sono stati ottenuti dagli ospedali in Russia e Vietnam in base alla IRB approvato protocolli e con IRB approvato moduli di consenso per l'analisi molecolare e genetica. I centri medici stessi hanno anche comitati etici interni con rivisto il protocollo e ICF. I campioni sono stati reperiti attraverso il tessuto Solutions Ltd http: //www. Tessutali soluzioni com /.. Per le caratteristiche dei campioni Cfr Tabella file 1 S1
Array
genotipi e copiare i profili numero sono stati generati per ogni campioni utilizzando 1 mg di corsa DNA sugli array Affymetrix SNP V6 utilizzando protocolli Affymetrix. numero di copie dei dati di variazione sono stati analizzati con il software http ArrayStudio: //www. Omicsoft com.. I dati sono stati normalizzati utilizzando l'algoritmo Affymetrix e segmentato con CBS. Un profilo trascrizione è stata generata per ogni campione utilizzando 1 mg di funzionamento totale RNA su Illumnia HG-12 array di espressione dell'RNA seguenti protocolli di Illumina. I dati sono stati analizzati con il software http Illumina GenomeStudio:.. //Www Illumina com /software /genomestudio_ software ILMN.. Come procedura di dati pre-elaborazione, un insieme sonda è stata mantenuta solo se ha un "presente" (cioè due deviazioni standard sopra sfondo) chiamare in almeno uno dei campioni. I valori dei segnali dei restanti set di sonde sono state trasformate in scala logaritmica 2-based ed è stata effettuata la normalizzazione quantile. copia del DNA e livelli di espressione di RNA sono stati integrati a livello del gene all'interno del software http ArrayStudio: //www. Omicsoft com.. analisi di arricchimento Pathway è stata effettuata all'interno dell'analisi metacore Suite http GeneGO: //www. genego com /.. Tutti i dati di matrice di questo studio è disponibile in GEO http: //www NCBI nlm NIH gov /geo /con il numero di serie adesione GSE29999
mirato il sequenziamento del DNA in profondità
5..... mg di DNA era PCR-arricchito per gli esoni codificanti di qualsiasi trascrizione conosciuta di 384 geni di interesse (file aggiuntivo 2 tabella S2) utilizzando la piattaforma http Raindance:... //www tecno- raindancetechnol com /
Le librerie bersaglio risultanti sono stati sequenziati usando Illumnia GAII ad una lunghezza lettura di 54 nt. Sequenza legge sono stati mappati al genoma di riferimento (hg18) utilizzando il programma BWA [44]. Basi di fuori delle regioni interessate sono stati ignorati quando riassume le statistiche di copertura e le chiamate variante. SAMtools è stato utilizzato per analizzare gli allineamenti ed effettuare chiamate genotipo [45], e qualsiasi chiamata che si discosta da base di riferimento è stato considerato come un potenziale variante. Il pacchetto SAMtools genera stime di qualità consenso e di qualità variante di caratterizzare le chiamate genotipo. La precisione delle chiamate genotipo è stato stimato da concordanza al genotipo chiamate dalla Affymetrix 6.0 SNP microarray. matrici di concordanza dei campioni sulla base di dati sia SNP e di sequenza sono stati generati per verificare la presenza di errori di etichettatura del campione (ulteriore file di 3 figura S1). Concordanza e la quantità di chiamate genotipo sono stati tabulati per le soglie di qualità del consenso, la qualità variante, e la profondità. L'insieme finale di chiamate varianti sono stati identificati usando qualità consenso maggiore o uguale a 50 e di qualità variante maggiore di 0. Per identificare esclusivamente modifiche somatiche, solo le mutazioni presenti nel campione cancro e non identificati in nessuno dei campioni normali sono stati mantenuti. Come un filtro aggiuntivo per le varianti della linea germinale, sono state rimosse tutte le varianti presenti in dbSNP e 1000 set di dati del genoma polimorfismo.
Q-PCR
Q-PCR è stata effettuata tramite protocollo standard utilizzando Fluidigm 48 * 48 array dinamico. In primo luogo, una corsa di validazione è stata condotta utilizzando pool di RNA di controllo di tre esemplari. Quattro quantità di RNA di ingresso sono stati testati (125 ng, 250 ng, 375 ng e 500 ng). punti dati triplicato sono stati ottenuti per la diluizione in seguito 10 punti di serie per ogni condizione di ogni test. I migliori risultati complessivi erano 250 o 500 ng, che ha prodotto valori di efficienza ~ 85%. Pertanto 250 quota di immissione ng per i campioni sperimentali. I dati è stato prodotto in triplice copia e significare combinato. valori CT sono stati convertiti in abbondanza con abbondanza formula standard = 10 (40-CT /3,5). dati di test è stato normalizzato per governanti utilizzando l'analisi del metodo di covarianza per cui i due governanti (GAPDH e beta-actina) sono stati usati per calcolare un punteggio solida e il punteggio è stato utilizzato come covariata per regolare gli altri geni. L'analisi dei dati è stata effettuata nel software Arraystudio.
sequenziamento Sanger
primer DNA genomico PCR sono stati ordinati da IDT (Integrated DNA Technologies Inc, Coralville, Iowa). reazioni di PCR sono state effettuate utilizzando Invitrogen Platnium polimerasi (Invitrogen, Carlsbad, CA). 50 ng di DNA genomico è stato amplificato per 35 cicli a 94 ° C per 30 secondi, 58 ° C per 30 secondi e 68 ° C per 45 secondi. I prodotti di PCR sono stati purificati mediante Agencourt AmPure (Agencourt Bioscience Corporation, Beverly, MA). sequenziamento diretto dei prodotti di PCR purificati con primer di sequenziamento sono stati effettuati con AB v3.1 BigDye-terminator kit ciclo di sequenziamento (Applied Biosystems, Foster City, CA) e le reazioni di sequenziamento sono stati purificati mediante Agencourt CleanSeq (Agencourt Bioscience Corporation, Beverly, MA). Le reazioni di sequenziamento sono stati analizzati utilizzando un Genetic Analyzer 3730XL (Applied Biosystems, Foster City, CA). Tutti i dati dei risultati della sequenza sono stati assemblati e analizzati utilizzando Codon Codice Aligner (CodonCode Corporation, Dedham, MA).
Risultati
DNA e amplificazione di RNA modelli attraverso i campioni sono coerenti con gli studi precedenti
In linea con la maggior parte degli altri tumori umani, del numero di copie variazioni intervenute tra il genoma dei campioni di cancro gastrico 50 rispetto ai campioni normali corrispondenti (Figura 1). Grandi regioni di amplificazione frequenti sono stati trovati in regioni cromosomiche 8Q, 13q, 20q e 20p. oncogeni MYC noti
e CCNE1
si trovano nelle 8Q e 20p amplificati, rispettivamente e probabilmente contribuiscono a un vantaggio di crescita conferito dal l'amplificazione. Queste amplificazioni sono stati osservati in studi precedenti in cancro gastrico con l'amplificazione di 20p per i quali ZNF217
e TNFRSF6B
sono stati suggeriti come i geni macchinista [46]. Figura 1 Vista aberrazioni CNV in tutti i 50 campioni di carcinoma gastrico, per ogni autosome. L'asse y corrisponde alla somma del numero di cambiamenti positivi o negativi per un particolare segmento con il rapporto log2 di quelli cambiamento. Le aree con il numero maggiore o minore copia coerente in tutti i campioni analizzati o molto grandi cambiamenti in alcuni campioni mostreranno grandi dimensioni positive e negative del cambiamento. Ogni punto o segmento in figura è colorato a campione. Il codice colore è arbitrario con ciascuno dei campioni 50 cancro essere assegnato un colore. segmenti amplificati includono 8q cromosoma, 20q, 20p, 3q, 7p, e 1Q.
concordanza tra numero di copie di DNA di guadagno e di espressione dell'RNA tra i campioni di cancro è stato valutato e le 200 geni contenuti in una regione di frequente copia ad alta DNA campioni di cancro e che avevano alti livelli di mRNA (rispetto al tessuto normale corrispondono) sono tabulati in di file 4 Tabella S3. La maggior parte dei geni presenti in questa lista sono da regioni cromosomiche 20q e 8Q, suggerendo che queste amplificazioni hanno più effetto sui livelli di mRNA, in minoranza sono geni per 20p, 3q, 7p, e 1q. La figura 2 mostra i profili di RNA misurati mediante Q-PCR di un gene esemplare da ogni regione mostrando sovraespressione generale nel cancro gastrico, soprattutto in alcune campioni. Oltre MYC
e CCNE1
, ci sono più geni in queste regioni, che potrebbero contribuire ad un vantaggio di crescita per la cellula tumorale. I percorsi biologici più significativamente arricchito da geni amplificati e sovraespressi sono coinvolti nella regolazione della traduzione (p = 0,000,015 mila) e danni al DNA di riparazione (p = 0,003). I campioni con amplificazioni in queste regioni genomiche sono annotati in Figura 3. Non vi è alcuna tendenza riconoscibile per amplificazioni in queste regioni di co-verificarsi o per essere esclusivo. In accordo con uno studio precedente [47], il PERLD1
locus è stato amplificato (all'interno del ERBB2
amplicone) nel campione di 08280 e MMP9
era sovraespresso, ma non visibilmente amplificato. Anche in Figura 3 sono indicati amplificazioni di DNA focali con l'espressione di RNA concorde di geni che possono influenzare la risposta alle terapie mirate, ad esempio dati sottostanti visualizzare ulteriori file di 5 figura S2. Figura 2 Espressione di esempi di geni provenienti da ogni regione cromosomica amplificato attraverso campioni di studio confermati da Q-PCR. I puntini rossi indicano i campioni tumorali e puntini bianchi denotano campioni normali. L'asse y indica l'abbondanza mRNA.
Figura 3 profilo mutazionale di campioni. Campioni di tessuto sono visualizzate nella parte superiore e le annotazioni rilevanti per loro sono in colonne sottostanti. Caselle rosse denotano l'amplificazione del DNA e concordanti sovraespressione di mRNA, scatole arancio denotano RNA sovraespressione senza evidenza di amplificazione del DNA, puntini rossi indicano la perdita del DNA. scatole blu denotano somatica mutazioni nonsynonymous convalidato da Sanger sequenziamento e scatole viola denotano mutazioni somatiche non sinonime, osservati nei dati Illumina con nessun tentativo di confermare con Sanger sequenziamento. modifiche aminoacidi sono noti nelle caselle e le modifiche che portano alla perdita o il guadagno di un codone di stop sono in rosso.
dati di sequenziamento mostra alta concordanza con genotipizzazione
Sequencing preparazione biblioteca non era riuscito per sei dei campioni originali 50 di cancro e quattordici dei campioni normali originali corrispondenti. Pertanto due coppie più abbinati stati aggiunti all'analisi, risultando in un set di dati di 44 campioni di tumore, 36 con coppie normali appaiati (ulteriori Tabella file 1 S1). La regione di destinazione incluso 3.28 MB tutto 6.547 esoni unici in 384 geni (ulteriore file di 2 tabella S2). una copertura media di tutti i campioni è stata 88,3% e sceso al 74% quando si richiede una copertura minima di 20. Tutti sequenziamento è stato effettuato per un minimo di 110x copertura media di lettura attraverso le regioni genomiche arricchiti per ogni campione. La legge sono stati allineati contro il genoma umano e varianti dal genoma di riferimento sono stati chiamati. Come controllo, un'analisi per confrontare la genotipizzazione chiamate da array Affymetrix V6 SNP e è stato eseguito il sequenziamento Illumina. Le regioni mirati per il sequenziamento contenevano 1.005 loci coperto dalle array Affymetrix V6 SNP. In assenza di filtraggio della variante sequenziamento richiede parametri di qualità, l'accordo tra la mediana genotipizzazione e sequenziamento risultati è stata del 97,8% con un range di 65-99% (addizionale 6a di file, Figura S3a). Il grezzo concordanza generale chiamata genotipo era 96,8%. metriche di qualità sono stati scelti per massimizzare l'accordo tra la genotipizzazione e le chiamate di sequenziamento, riducendo al minimo i falsi negativi. La metrica più informativo era la qualità del consenso e un cut-off di ≥50 comportato la perdita di circa il 10% dei genotipi condivisi, ma un aumento complessivo del 2% in concordanza al 98,7% (supplementare 6b di file, Figura S3b). chiamate genotipo Variant sono stati isolati per ulteriori analisi concordanza. In questo insieme, una soglia di qualità variante > 0 maggiore precisione di chiamate genotipo variante al 98,9% (addizionale 6c di file, Figura S3c). Quando entrambe le soglie di qualità sono stati applicati la concordanza campione mediana è del 99,5% (addizionale 6d di file, figura S3D) che è all'interno della regione di errore di allineamento genotipizzazione. Sei campioni (08362T1, 08373T2, 336MHAXA, 08337T1, 89362T2, DV41BNOH) hanno avuto una concordanza di < 98% e due di questi (08393T2 e DV41BNOH) hanno rispettivamente una concordanza del 82% e 88%. Quindi con un ≥ qualità di consenso 50 e una qualità variante > 0, la percentuale di falsi positivi è stato dello 0,5% e del 1,6% per i genotipi di riferimento e genotipi variante, rispettivamente (file aggiuntivo 6e figura S3E).
Da tutti single nucleotide cambiamenti che passano le soglie di cui sopra, tutte le varianti presenti in nessuno dei campioni normali o nei database di polimorfismo dbSNP (V130) o 1000 genomi sono stati assunti varianti linea germinale e scartato. Varianti presenti solo negli esoni di campioni tumorali sono stati assunti per essere somatica e conservati. 18,549 varianti somatici sono stati rilevati in totale in tutti i 44 campioni (file aggiuntivo 7 Tabella S4), 3357 sono stati previsti per essere exonic e non sinonime. Per assegnare una priorità per le mutazioni con impatto funzionale ci concentriamo tutte le ulteriori analisi sulle mutazioni non sinonime e le mutazioni evidenziati portano alla perdita o il guadagno di codoni di stop. Abbiamo applicato l'algoritmo SIFT [48] per prevedere i cambiamenti di aminoacidi che non sono tollerati in evoluzione e quindi hanno più probabilità di influenzare la funzione della proteina, 1509 mutazioni somatiche non sinonime hanno un punteggio SIFT di < 0.05. Il tasso di mutazioni con SIFT score < 0,05 per gene, corretto per la lunghezza CDS è stato calcolato (4). La figura 4 mostra, i geni con la più alta concentrazione di bassa SIFT mutazioni segnando erano S1PR2
, LPAR2
, SSTR1
, TP53
, GPR78
e RET
, con S1PR2 essendo più estremo. Ci sono quindici mutazioni con SIFT score < 0.05 in tutto il 353aa CDS di S1PR2
, concentrati in nove campioni. S1PR2
anche conosciuto come EDG5 Codici promozionali per un recettore accoppiato a proteine G di S1P e attiva RhoGEF, LARG
[49]. Poco si sa del suo ruolo nel cancro e mutazioni somatiche non sono stati osservati in 44 tessuti sequenziato per S1PR2
nel database COSMIC [50]. Figura 4 Bar grafico della frequenza di mutazioni deleterie attraverso gene sequenziato. I geni in sequenza vengono visualizzati sul asse x. Il numero di mutazioni deleterie non sinonime somatiche osservate in ogni gene /numero di aminoacidi in ogni CDS in tracciati.
Dati di sequenziamento è confermato dal sequenziamento Sanger
Alcune mutazioni somatiche non sinonime sono stati selezionati per essere confermata dal sequenziamento Sanger. Tutte le mutazioni riportate in blu nella figura 3 sono stati confermati da Sanger sequenziamento e sono stati confermati anche per essere somatica mediante sequenziamento della sequenza di tipo selvatico nel tessuto normale abbinato (vedi ulteriore file di 8 Figura S4 per esempio, tracce di sequenziamento). Anche se il 74% sono state confermate, alcune mutazioni rilevate nel sequenziamento Illumnia non sono stati confermati come mutazioni somatiche di sequenziamento Sanger. Sedici dei 68 (24%) mutazioni abbiamo tentato di confermare erano presenti nel campione normale e il cancro, questi sono mutazioni germinali ma non rilevati in nessuno dei campioni normali da Illumina sequenziamento e, inoltre, non rappresentati nel 1000 i dati genomi dbSNP o. Cinque dei sedici mutazioni germinali erano da campioni tumorali senza tessuto normale abbinato incluso nel set di dati, gli altri undici provenienti da campioni tumorali con abbinato normale sequenza tessuto incluso nel set di dati. Ciò evidenzia un tasso di contaminazione linea germinale non eliminate dai controlli normali appaiati o il confronto di database polimorfismo noti. Può essere che la copertura delle sostituzioni nel tessuto normale sembra essere inferiore rispetto al campione di cancro e così alcune mutazioni della linea germinale restano nonostante i filtri somatiche. Due dei 68 (3%) mutazioni abbiamo tentato di confermare non erano presenti nel campione normale o il cancro per Sanger sequenziamento. Una causa potrebbe essere falsi positivi nei dati Illumnia a causa di manufatto; tuttavia file aggiuntivo 6 Figura S3 mostra la percentuale di falsi positivi sia bassa almeno per quelle varianti rappresentate sulle array Affymetrix V6. Un'altra possibilità è che questi sono presenti in un sottoinsieme del campione inferiore alla sensibilità del metodo Sanger ma rilevato dal sequenziamento Illumina. Pertanto, le mutazioni riportati nel sequenziamento Illumina sono riportati anche in viola nella figura 3, una certa cautela è giustificata quando si interpretano questi risultati in quanto potrebbero essere polimorfismi germinali o presenti solo in un sottoinsieme del campione di tumore.
Alterazioni nel RAS /RAF /MEK /ERK
campioni Tre tumore KRAS avevano
alterazioni genetiche (Figura 3), suggerendo opportunità terapeutica per il trattamento con inibitori della MEK. Una di queste alterazioni è una mutazione G12D. KRAS
mutazioni G12D hanno dimostrato di iniziare la cancerogenesi e la sopravvivenza del tumore [51]. Amplificazione e sovraespressione di di tipo selvatico KRAS
è stato visto negli altri 2 campioni. KRAS
di amplificazione è stato osservato prima nel 5% dei tumori gastrici primari. linee cellulari di cancro gastrico con di tipo selvatico KRAS
amplificazione Mostra KRAS costitutivi
attivazione e la sensibilità di KRAS
RNAi atterramento [24]. è stata anche osservata una nuova mutazione nel gene KRAS
; . (Nel campione di 08393) la conseguenza funzionale è sconosciuto
Il PIK3CA
mutazione co-occorrenti con KRAS G12D
, è noto per influenzare la sensibilità agli inibitori MEK [25]; Inoltre, nuove mutazioni osservati in questo studio possono anche avere conseguenze per la stessa classe terapeutica. Per esempio: KSR2
funge da impalcatura molecolare per promuovere ERK segnalazione [52, 53]. Pertanto, le mutazioni in KSR2
come visto in sette campioni possono influenzare la sensibilità agli inibitori MEK. Un secondo esempio è ULK1
, che controlla positivamente autofagia valle di mTOR [54] ed è mutato in quattordici campioni. L'autofagia è aumentata insieme a ERK fosforilazione quando le cellule di cancro gastrico sono trattati con un inibitore del proteasoma [55], quindi mutazioni in ULK1
possono influenzare la sensibilità ai trattamenti inibitori del proteasoma, come bortezomib in monoterapia o in combinazione con gli inibitori della MEK.
alterazioni nella via PI3K /AKT
C'era notevole sequenza di interruzione delle phosphoinositide-3-chinasi (PI3K) geni pathway a campionario. Ci sono una serie di PI3K /AKT /mTOR inibitori in sviluppo clinico e nei pazienti con mutazioni attivanti nel percorso sono candidati per il trattamento [56]. PIK3CA
mutazioni del oncogenicità noto sono stati trovati in quattro campioni. Ciò si traduce in una frequenza di PIK3CA
mutazione hotspot del 9%, leggermente superiore rispetto alle precedenti stime di 6% (12/185) [27] e il 4,3% (4/94) [57]. L'hotspot mutazioni comune PIK3CA di oncogenesi nota (E545K e H1047R) [58] sono state osservate due volte. Un'altra mutazione in PIK3CA
K111E, che è stata anche osservata prima in quattro campioni in COSMIC, è stata osservata una sola volta e potenzialmente nuove mutazioni somatiche sono state osservate in altri due campioni. Sono stati osservati
Cinque nonsynonymous AKT1
mutazioni. Anche se Akt1
mutazioni sono presenti in circa il 2% di tutti i tumori, si verificano principalmente a aminoacidi 15 e l'importanza funzionale della mutazione in altri siti è sconosciuta. Un'altra mutazione nonsynonymous in AKT2
è stata osservata nel campione 08407. Akt2
mutazioni sono molto più rari rispetto AKT1
mutazioni, anche se un AKT2
mutazione è stata osservata prima nel carcinoma gastrico, con una frequenza 2% [ ,,,0],59]. Infine la mutazione di PTEN
o MTOR
possono influenzare la risposta al percorso inibitori. Alterazioni Diversi PTEN
mutazioni sono notati e mTOR
mutazioni sono frequenti.
In recettore tirosin chinasi
recettore delle tirosin chinasi (RTK) e di droga obiettivi EGFR
, ERBB2
e MET
sono stati ogni amplificato (log 2 > 0,6) e sovraespresso a livello di RNA in un campione di cancro. Ne consegue che i tumori possono essere sensibili agli inibitori dei RTK amplificati. Inoltre, mutazioni multiple non sinonime si osservano nelle loro regioni codificanti. mutazioni a valle ci si aspetterebbe di influenzare la risposta. Ad esempio, nel MET
campione amplificato una mutazione troncando in Akt3
può influenzare la sensibilità agli inibitori MET.
FGFR2
viene amplificato e RNA sovraespresso in due campioni, ci sono anche diverse mutazioni in FGFR1
-4. Gamma inibitori di massima per RTK, che prendono di mira FGFRs tra le altre chinasi, possono essere efficaci in questi pazienti [60, 61].
Alterazioni nella cella proteine del ciclo
Il viral oncogene omologo SRC
è mutato in quattro del tumore campioni, due delle mutazioni sono previsti per avere un effetto deleterio compresa l'introduzione di un codone di stop. Questo può contrattaccare indicare inibitori SRC. MET
amplificazione è anche un marcatore di resistenza noto per le terapie anti-SRC come dasatanib [62, 63]. La chinasi del ciclo cellulare connessi, AURKA
è stato amplificato e sovraespresso in un campione. Gli inibitori AURKA sono in fase di sviluppo per i tumori solidi [37] e possono essere indicati in questo caso. CCNE1
è stato amplificato in due campioni (08390 e 08357). Alti livelli di CCNE1
hanno dimostrato di essere frequentemente associate a precoce del cancro gastrico ed i livelli di metastasi ma l'espressione non correlano con la sopravvivenza [64, 65]. Alti livelli di CCNE1
sono stati proposti come marker della sensibilità per i pro-farmaco terapie enzimi attivati gene-diretto [66]
attivazione del pathway Wnt è comune nei campioni di carcinoma
Le mutazioni sono stati osservati nel APC
gene in 22 campioni. APC è un soppressore del tumore nota per attivare CTNNB1 e Wnt percorso di segnalazione, tra gli altri effetti [67]. La via di Wnt è stato precedentemente trovato ad essere frequentemente attivati in cancro gastrico [68]. Abbiamo usato una firma trascrizionale, generato da studi precedenti [69, 70] e sono disponibili presso la banca dati Broad Institute MSigDB per classificare i campioni di studio con le loro firme Wnt trascrizionale. La figura 5A mostra una mappa di calore dei livelli trascrizionali dei geni firma WNT nei set di dati. L'attivazione di questo percorso è maggiore in quasi tutti i campioni tumorali rispetto ai campioni normali. Gli inibitori Wnt sono oggetto di intensa ricerca nel settore farmaceutico e della ricerca accademica [71-73]. Questi risultati suggeriscono che avranno l'indicazione nel carcinoma gastrico, così come molti altri tipi di tumore. Figura 5 firme trascrizionali attraverso campioni. heatmap cluster mostrando espressione di un Wnt geni firma e B geni firma riccio, attraverso campioni nello studio. Tutti i valori di espressione sono Zscore normalizzati. Zscore < -1 sono blu, Z-score > 1 sono di colore rosso con una colorazione classificato attraverso bianco a 0. nomi dei campioni sono l'asse x, si sono raggruppati per modello di espressione e campioni con punteggi elevati di firma sono a destra. I campioni con mutazioni somatiche non sinonime APC (A) o mutazioni Ptch1 (B) e contrassegnata da un asterisco sopra la heatmaps. WNT firma geni (dall'alto in basso): FSTL1, DACT1, CD99, LMNA, SERPINE1, TNFAIP3, GNAI2, ID2, MVP, ACTN4, CAPN1, LUZP1, MTA1, RPS19, PTPRE, AXIN2, NKD2, SFRS6, CCND1, SCAP, CPSF4 , SENP2, DKK1, PRKCSH, SLC1A5, HDGF, CBX3, SCML1, PCNA, RPS11, SNRPA1, TGM2, LY6E, IFITM1, NSMAF, TCF20, BCAP31, AXIN1, Agrò, PLEKHA1, SLC2A1, CTNNB1, EIF5A, IMPDH2, GSK3B, PFN1 , UBE, MAP3K11, ARHGDIA, HNRPUL1, FLOT2, GYPC, NCOA3, CENTB1, SYK, POLR2A, KRT5, DHX36, ELF1, SMG2, FGD6, MAPKAP1, LOC389435, RPL27A, SRP19, RPL39L, SFRS2IP, FUSIP1
; Hedgehog firma geni (dall'alto in basso):. LRFN4, JAG2, RPL29, Wnt5a, SNAI2, FST, MYCN, BMP4, CCND1, BMI1, CFLAR, PRDM1, GREM1, FOXF1, CCND2, CD44
attivazione del riccio percorso è comune anche nei carcinoma campioni
PTCH1
è un soppressore del tumore e agisce come un recettore per i ligandi riccio e inibisce la funzione di lisciato. Quando lisciato viene liberato, segnala intracellulare che porta all'attivazione di fattori di trascrizione GLI [74]. Molteplici mutazioni somatiche di PTCH1 Quali sono registrati nel COSMIC, in linea con il suo ruolo di soppressore del tumore.
 Rifornimento dell'armadio del negozio SIBO – Parte 2
Allora, come sta andando lo stoccaggio del tuo armadio per negozi SIBO? Hai pulito la primavera o organizzato lautunno i tuoi armadi per il cibo e ti sei sbarazzato di cose vecchie e allettanti, cose
Rifornimento dell'armadio del negozio SIBO – Parte 2
Allora, come sta andando lo stoccaggio del tuo armadio per negozi SIBO? Hai pulito la primavera o organizzato lautunno i tuoi armadi per il cibo e ti sei sbarazzato di cose vecchie e allettanti, cose
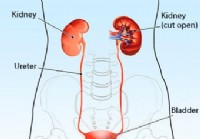 Infezione renale (pielonefrite)
Illustrazione dei reni, delluretere, della vescica e delluretra. Definizione e fatti dellinfezione renale Linfezione renale è una delle numerose infezioni che possono coinvolgere le vie urinarie. Lin
Infezione renale (pielonefrite)
Illustrazione dei reni, delluretere, della vescica e delluretra. Definizione e fatti dellinfezione renale Linfezione renale è una delle numerose infezioni che possono coinvolgere le vie urinarie. Lin
 La differenza tra buoni e grandi praticanti (mi ha salvato la vita)
Ho sprecato QUANTI soldi per rimettermi in salute? Questa domanda mi ha infastidito per mesi, soprattutto quando ho pensato alle mie bollette. Così ho iniziato a controllare le mie carte di credito
La differenza tra buoni e grandi praticanti (mi ha salvato la vita)
Ho sprecato QUANTI soldi per rimettermi in salute? Questa domanda mi ha infastidito per mesi, soprattutto quando ho pensato alle mie bollette. Così ho iniziato a controllare le mie carte di credito