Résumé
Beaucoup de cancers solides sont connus pour présenter un degré élevé d'hétérogénéité dans leur dérégulation des différentes voies oncogéniques. Nous avons cherché à identifier les principales voies oncogéniques dans le cancer gastrique (GC) avec des relations importantes à la survie des patients. Utilisation des signatures d'expression génique, nous avons conçu un in silico Le cancer gastrique est le deuxième cause de mortalité dans le monde du cancer. Avec les traitements actuels, moins d'un quart des patients survivent plus de cinq ans après la chirurgie. cancers gastriques individuels sont très disparates dans leurs caractéristiques cellulaires et les réponses aux médicaments chimiothérapeutiques standard, ce qui cancer de l'estomac une maladie complexe. Pathway approches fondées, plutôt que des études monogéniques, peuvent aider à démêler cette complexité. Ici, nous utilisons une approche de calcul pour identifier les connexions entre les voies moléculaires et les profils de cancer. Dans une étude à grande échelle de plus de 300 patients, nous avons identifié des sous-groupes de cancers gastriques distinguables par leurs habitudes de conduite des voies moléculaires. Nous montrons que ces sous-groupes identifiés sont cliniquement pertinentes pour prédire la durée de survie et peuvent se révéler utiles pour guider le choix des thérapies ciblées visant à interférer avec ces voies moléculaires. Nous avons également identifié des lignées de cellules de cancer gastrique spécifiques reflétant ces sous-groupes de la voie, ce qui devrait faciliter l'évaluation pré-clinique des réponses aux thérapies ciblées dans chaque sous-groupe Citation:. Ooi CH, Ivanova T, Wu J, Lee M, Tan IB, Tao J, et al. (2009) oncogènes Pathway Combinaisons Prédire pronostic clinique dans le cancer gastrique. PLoS Genet 5 (10): e1000676. doi: 10.1371 /journal.pgen.1000676 Editeur: Jason G. Mezey, Cornell University, États-Unis d'Amérique Reçu le 22 Avril 2009; Accepté: 3 Septembre 2009; Publié 2 Octobre, 2009 Droit d'auteur: © 2009 Ooi et al. Ceci est un article en accès libre distribué sous les termes de la licence Creative Commons Attribution, qui permet une utilisation sans restriction, la distribution et la reproduction sur tout support, à condition que l'auteur et la source originelle sont crédités Financement:. Ce travail a été soutenue par des subventions à PT de BMRC 05/1/31/19/423, Singapour Cancer Syndicate SCS-BS0001, NMRC accorder TCR /001/2007, et un noyau subvention Duke-NUS. Les bailleurs de fonds ont joué aucun rôle dans la conception de l'étude, la collecte et l'analyse des données, la décision de publier, ou de la préparation du manuscrit Intérêts concurrents:.. Les auteurs ont déclaré aucun conflit d'intérêts existent Introduction le cancer gastrique (GC) est la deuxième cause de mortalité par cancer dans le monde [1]. Particulièrement répandue en Asie, la plupart des patients du GC sont diagnostiqués avec la maladie à un stade avancé [2]. Déréglementation des voies oncogéniques canoniques tels que E2F, K-RAS, p53, et la signalisation Wnt /β-caténine sont connus pour se produire avec des fréquences variables dans GC [3] - [6], indiquant que GC est une maladie hétérogène moléculairement. Des études antérieures décrivant la diversité de GC dans les tumeurs primaires ont généralement porté sur des voies simples, mesurant seulement un ou quelques biomarqueurs par expérience [4], [6], [7]. En revanche, les données expérimentales indiquent que la plupart des phénotypes de cancer (croissance incontrôlée, la résistance à l'apoptose, etc.) sont en grande partie régies non seulement par des voies simples, mais les interactions complexes entre les pro- multiples et des circuits de signalisation anti-oncogènes [8]. Combler cet écart entre les arènes cliniques et expérimentales nécessiteront des stratégies capables de mesurer et concernant l'activité des motifs de multiples voies oncogéniques simultanément dans les tumeurs primaires. Des études antérieures ont proposé d'utiliser des signatures d'expression génique pour prédire l'activité des voies oncogéniques dans cancers [9] - ici, nous avons émis l'hypothèse que les schémas d'activation de la voie oncogénique pourraient être utilisés pour développer une taxonomie génomique du GC. Fait important, cette stratégie de voie centrée diffère sensiblement des études de microréseaux antérieures décrivant les changements d'expression associés aux différences de type morphologique et de tissus en GC [10], [11], que les signatures de la voie (plutôt que des gènes individuels) sont utilisés comme base pour la classification du cancer . Nous avons développé un in silico Résultats Prédiction Pathway Activation en Cancer Gene Expression Profils Notre stratégie pour prédire les niveaux d'activation de la voie oncogénique dans les cancers comporte quatre étapes (figure 1A). Tout d'abord, nous avons défini «signatures de la voie» - ensembles de gènes présentant une expression altérée après perturbation fonctionnelle d'une voie spécifique dans un in vitro Avant d'appliquer cette approche à la GC, nous avons considéré qu'il était important de valider ce in silico Deuxièmement, nous avons testé si une signature de la voie de signalisation associée à l'œstrogène, mais dérivé de non le tissu mammaire peut également être utilisé pour stratifier le même panel de lignées cellulaires de cancer du sein. Nous avons demandé le panneau de la lignée cellulaire de cancer du sein avec un 41-gène "réponse œstrogène" signature dérivée d'une liste de gènes surexprimés par l'estradiol dans U2OS cellules d'ostéosarcome humain [20]. En dépit de la signature en provenance d'un autre type de tissu (par exemple, un ostéosarcome), on a trouvé à nouveau que, lorsqu'ils sont triés en fonction de leur prédit la réactivité des oestrogènes, des lignées cellulaires de cancer du sein regroupés selon leur niveau de (le récepteur d'oestrogène) de ESR1 expression (p = 0,0035, Exactitude 62,7%, 94,7% Sensibilité, Spécificité 43,8%) (figure 1C et tableau S1). Ces résultats démontrent qu'il est en effet possible de prédire les tendances de l'activation de la voie dans un cancer d'intérêt particulier (cancer gastrique dans nos cas) en utilisant des signatures d'expression obtenus à partir de différentes conditions expérimentales et les types de tissus, même différents. Après avoir validé cette approche de prédiction de la voie, nous avons procédé à appliquer la stratégie de GC primaire. Plutôt que de tester chaque voie possible, nous avons sélectionné onze voies de suppression oncogènes et tumorales précédemment impliqués dans la carcinogenèse gastrique, en utilisant dans nos RAS d'analyse [4], p53 [5], BRCA1 [12], p21 [13], Wnt /β-caténine [6], E2F [3], SRC [14], MYC [15], NF-kB [21], la désacétylation des histones (HDAC) [16], et les cellules souches liées à des signatures [17]. Chaque fois que possible, nous avons tenté de sélectionner plusieurs signatures pour chaque voie, de préférence à partir d'études publiées indépendantes. Par exemple, des deux signatures d'activation E2F utilisés dans notre approche, une signature a été obtenue en induisant l'activité E2F1 dans les cellules de fibroblastes de rat [22] tandis que l'autre signature a été obtenue en utilisant une lignée de cellules d'ostéosarcome dérivés contenant une protéine de fusion ER-E2F1 inductible [23]. prédictions de la voie finale pour d'autres analyses ont été généralement obtenues en combinant signatures individuelles appartenant à la même voie (voir Matériels et Méthodes). Nous avons calculé les scores d'activation pour les onze voies représentées par 20 signatures de la voie sur trois cohortes indépendantes de primaire GCS dérivé de l'Australie (cohorte 1-70 tumeurs), Singapour (cohorte 2-200 tumeurs), et le Royaume-Uni (cohorte 3-31 tumeurs). Pour visualiser les modèles d'activation de la voie, nous dépeints chaque cohorte comme heatmap, où la couleur de heatmap représente la force prédite d'activation pour chaque voie dans l'individu GCS. Nous avons observé une hétérogénéité considérable d'activation de la voie entre les patients du GC individuels (Figure 2A-2C). Cependant, les signatures provenant d'études indépendantes représentant des voies semblables ont donné souvent des modèles de prédiction similaires (par exemple NF-kB (peau) et NF-kB (col)), et un test de chi-carré a confirmé un niveau significatif de similitude dans les modèles généraux de voie activation entre les cohortes Australie et Singapour (p = 0,00038), et entre les cohortes Australie et au Royaume-Uni (p = 0,00051, voir le tableau S2) suggérant que les prédictions de la voie de GC ne sont pas liés à une cohorte de patients spécifiques. Nous avons identifié deux grands groupes de voies de co-actif, qui ont été complètement conservés dans Cohortes 1 et 2 (figure 2A et 2B) et la plupart conservés dans la cohorte 3 (figure 2C). Ceux-ci inclus (i) une «prolifération /cellules souches« cluster de voie (barre verticale brune à la figure 2) englobant les voies associées aux différents régulateurs du cycle cellulaire (par exemple MYC, E2F, p21) et la tige signatures de cellules; et (ii) un cluster 'de signalisation oncogénique »de la voie (barre verticale grise Figure 2) contenant beaucoup de différentes voies oncogéniques (BRCA1, NF-kB, p53, Wnt /β-caténine, voies SRC, RAS et HDAC). In vitro en analysant la heatmap de la voie de GC dans la figure 2, nous avons sélectionné trois voies oncogéniques (NF-kB, Wnt /β-caténine, et la prolifération Pour valider expérimentalement ces prédictions de la voie de GC primaire, nous avons appliqué l'algorithme de prédiction de la voie à un panel de lignées cellulaires 25 GC (de GCCLs) ( Figure 3). Semblable à GC primaire, «prolifération /cellules souches" et "signalisation oncogénique" grappes de la voie ont également été observées dans les GCCLs. En outre, les signatures représentant la même voie, mais obtenue à partir de différentes études, comme les deux signatures de MYC dérivés indépendants [9], [24] également regroupés dans les lignées cellulaires de GC après regroupement hiérarchique non supervisé (entre parenthèses pourpres dans la figure 3). Guidée par les prédictions de la voie, nous avons identifié des lignées de cellules GC spécifiques présentant des modèles d'activité de la voie oncogénique miroir GCS primaire. La confiance dans la sélection de lignées cellulaires spécifiques comme in vitro Models in a été également obtenue en répétant la procédure de prédiction sept fois en utilisant une variété de profils de référence, allant du profil GCCL médiane des profils indépendants tels que la normale non malignes profils d'estomac (voir Matériels et Méthodes et Tableau S4). Des comparaisons par paires ont confirmé que les profils de référence des deux étaient plus susceptibles de produire des prédictions concordante de la voie que des prédictions contradictoires (Texte S1 et Tableau S4). Quelques exemples de lignes représentatives comprennent AZ521 et MKN28 cellules, qui activation d'exposition de la prolifération /tige voies cellulaires, les cellules YCC3 et AGS pour les voies Wnt /β-caténine et MKN1 et cellules SNU5 pour la voie NF-kB. d'abord, nous avons mesuré directement les taux de prolifération des 22 GCCLs et corrélé les données sur les taux de prolifération avec le score d'activation moyen de signatures dans la prolifération /tige cluster voie cellulaire. Il y avait une association significative entre les taux prolifératives déterminées expérimentalement et les scores d'activation de la voie (R = 0,4688, p = 0,0278) (figure 4A). Soutenir l'idée que les signatures de la voie oncogéniques sont des prédicteurs supérieurs de l'activité de la voie par rapport à l'expression des gènes de la voie clés simples, aucune association significative n'a été observée pour les deux MYC ou l'expression de E2F1 (p = 0,48 et 0,38 pour MYC et E2F1, respectivement) (figure S1 ). Deuxièmement, afin de valider les prédictions de la voie Wnt /β-caténine, nous avons analysé l'expression des différents composants de la voie Wnt (β-caténine, TCF4) et les niveaux relatifs de TCF /LEF activité transcriptionnelle en GC lignées cellulaires prévus pour être Wnt /β activé-catenin- ou Wnt /β-caténine-non activé. Sur les sept lignées cellulaires sélectionnées pour leur tractability expérimentale (par exemple, la facilité de transfection et les conditions de croissance convenables), nous avons trouvé que les deux β-caténine et le TCF /LEF facteur de transcription TCF4 (également connu sous le nom TCF7L2), les principaux composants de la voie de signalisation Wnt, ont été exprimées dans des lignées cellulaires GC prédits par l'activation de la voie des analyses pour avoir une activité élevée Wnt /β-caténine (AGS YCC3, Kato III et NCI-N87), mais ne sont pas exprimés dans les lignées deux sur trois (de SNU1 et SNU5) associés avec des scores d'activation de Wnt /ß-catenine incohérents ou faibles (figure 4B). En outre, afin de doser directement l'activité de la voie Wnt, nous avons déterminé TCF /LEF activité de transcription dans les lignées cellulaires par GC en utilisant Topflash, un plasmide exprimant la luciférase contenant des sites de liaison multimérisées THC. Le dosage Topflash confirmé haute TCF /LEF activité transcriptionnelle dans trois des quatre lignées cellulaires GC supposé avoir une haute Wnt activité /β-caténine (AGS YCC3, et Kato III), mais peu ou pas d'activité Topflash dans des lignées cellulaires GC associée à Wnt /β-caténine scores d'activation incohérentes ou faibles (SNU1, SNU5 et SNU16). En outre, les scores d'activation voie β-caténine étaient significativement plus élevés chez GCCLs avec plus de deux fois TCF /LEF activité transcriptionnelle (AGS, YCC3, Kato III, et NCI-N87) que dans GCCLs avec inférieure TCF /LEF activité transcriptionnelle (p = 0,007, figure 4B). Lorsque comparés aux gènes uniques, des associations supérieures à TCF /LEF activité transcriptionnelle ont été à nouveau observée en utilisant le score d'activation moyenne de Wnt signatures /β-caténine par rapport à soit β-caténine ou TCF4 (aka TCF7L2) expression seul (p = 0,038 pour les signatures vs p = 0,31 et 0,58 pour les β-caténine et TCF4, respectivement) (figure S1). Troisièmement, pour valider les prédictions de la voie NF-kB, nous avons sélectionné 11 GCCLs toujours prédit que soit NF-κB- activé ( «NF-kB /on", six GCCLs) ou NF-kB non activé ( «NF-kB /off», cinq GCCLs) (figure S2). l'expression génique accrue de p50 et p65, les sous-unités hétérodimères de NF-kB, on a observé dans la norme NF-kB /sur des lignées cellulaires GC par rapport à la norme NF-kB activation /désactivation des lignées cellulaires GC (p = 0,0002 pour la p50, p = 0,046 pour la p65, la figure 4C) et à l'expression de p65 au niveau de la protéine a été observée principalement dans le NF-kB /sur des lignes (figure 4C). En utilisant une immunocytochimie sur la paraffine lignées cellulaires GC fixés au formol, l'expression de la protéine p65 était plus fréquemment observée dans la norme NF-kB /sur des lignées cellulaires GC par rapport à la norme NF-kB activation /désactivation GC lignées cellulaires en termes de sous-localisation nucléaire, les pourcentages de cellules présentant une coloration (soit intensité nucléaire ou cytoplasmique), et la coloration (Tableau S5, Figure S3). Pour déterminer si le NF-kB /sur des lignées cellulaires GC présentaient également l'expression différentielle des gènes p65 régulé par rapport à la norme NF-kB activation /désactivation des lignées cellulaires GC, on a combiné la liste des gènes directement liés par le facteur p65 de transcription [25] avec des listes de les gènes régulés au niveau de l'ARNm par le TNF-α [26], un inducteur connu de l'activation de NF-kB. Utilisation de Gene Set Analysis Enrichment (GSEA, [27]), nous avons trouvé que les gènes cibles de p65 surexprimés par traitement TNF-α ont été significativement surexprimé dans NF-kB /sur des lignées cellulaires de GC par rapport à la norme NF-kB /off des lignées de cellules de GC (enrichissement normalisé score, NDA = 1.86; taux de fausses découvertes, FDR < 0,001, en bas panneau le plus, la figure 4C). A l'inverse, les gènes cibles p65 réprimés par le TNF-α ont été significativement sous-exprimés dans la norme NF-kB /sur des lignées cellulaires GC par rapport à la norme NF-kB /off des lignées cellulaires GC (NDA = -1,56, FDR = 0,019, plus en bas panneau, la figure 4C). Enfin, pour confirmer directement la présence d'une activité de NF-kB élevée, nous avons transfecté trois NF-kB /sur des lignées cellulaires GC et deux NF-kB activation /désactivation des lignées cellulaires GC avec un reporter de luciférase contenant un gène rapporteur NF-kB. Comme cela est représenté sur la figure 4D, les trois NF-kB /sur des lignées cellulaires de GC présentait une activité transcriptionnelle de NF-kB élevée par rapport aux deux lignes de NF-kB activation /désactivation GC cellulaires (p = 0,0084). pris collectivement, ces résultats soutiennent le concept que in silico Combinaisons Pathway Prédire cancer gastrique de survie des patients pour évaluer la pertinence clinique des sous-groupes de la voie identifiés, nous avons étudié si les modèles de voie co-activation comme illustré dans les heatmaps des différentes cohortes pourraient être liées à la survie des patients. Nous avons utilisé les données de survie globale de la cohorte 1 et cohorte 2 et patients stratifiés par leurs motifs prévus de l'activation de la voie. Un profil de GC primaire a été défini comme montrant le niveau d'activation élevé d'une voie lorsque le score d'activation est supérieure à zéro - à savoir être positivement associé à la signature de la voie. Les groupes de patients stratifiés soit par la tige /activation de la voie de prolifération cellulaire marquer seul ou le score d'activation de la voie NF-kB seul ne diffèrent pas significativement quant à leur survie globale (p > 0,05 pour la prolifération /cellules souches et la NF-kB dans les deux cohortes, la figure 5A et 5B). Toutefois, lorsque les scores d'activation de la voie ont été combinées, les patients avec des niveaux d'activation élevés de NF-kB et la prolifération /tige voies cellulaires avaient survie significativement plus courte par rapport aux patients ayant un faible niveau d'activation des deux NF-kB et la prolifération /tige voies cellulaires (p = 0,0399 et p = 0,0109 pour cohortes 1 et 2 respectivement, la figure 5D). activation de la voie /β-caténine était significativement associée à la survie des patients dans la cohorte 1, (p = 0,0056, figure 5C) mais pas dans la cohorte 2 (p = 0,0693, figure 5C). Cependant, les patients dans Cohortes 1 et 2 avec des niveaux d'activation élevés de Wnt /β-caténine et de la prolifération /tige voies cellulaires avaient survie significativement pire par rapport aux patients ayant des niveaux d'activation faibles des deux voies (p = 0,0073 et p = 0,0086, Figure 5E ). Pour référence les contributions des combinaisons de la voie par rapport aux critères histopathologiques connus, nous avons effectué une analyse multivariée, y compris les prévisions de la voie combinées et le stade tumoral pathologique (classification TNM: stades 1-4), le facteur pronostique le plus important dans la GC [28]. Dans les deux cohortes, l'activation combinée de la prolifération /cellules souches et les voies NF-kB se sont avérées être un facteur pronostique indépendant du stade de la tumeur (p = 0,003 et 0,048 pour Cohortes 1 et 2, respectivement) (tableau S6). De même, l'activation combinée de la prolifération /tige voies cellulaires et Wnt /β-caténine était un facteur pronostique indépendant dans la cohorte 1 et atteint une signification limite dans la cohorte 2 (p < 0,001 et p = 0,058, Tableau S7). Ces résultats démontrent que l'évaluation de l'état d'activation de la voie combinée est cliniquement pertinente et d'ailleurs peut fournir une information pronostique supplémentaire par rapport à l'étalon-or actuel de prédiction du pronostic du patient, la mise en scène de la tumeur TNM basée. Dans cette étude, nous avons cherché à subdiviser GCS en sous-groupes homogènes moléculairement comme une première étape pour individualiser les traitements des patients et l'amélioration des résultats. Il est important, à la différence des études précédentes GC microarray concernant les profils d'expression génique à l'histologie ou de type anatomique [10], [11], nous avons choisi de fonder nos subdivisions de GC sur les modèles d'activité de la voie oncogène. Après l'élaboration et la validation de cette nouvelle approche de classification, nous avons été en mesure de décrire, pour la première fois, une taxonomie génomique de GC basée sur des modèles d'activité de la voie oncogène. Notre approche est particulièrement appropriée pour des microarrays d'expression génique, car ces plates-formes interroger des milliers de transcrits d'ARNm dans chaque échantillon, ce qui permet l'évaluation de multiples voies simultanément dans une seule expérience. En revanche, une telle approche est actuellement pas possible au niveau de la protéine en raison du manque de plates-formes appropriées. En utilisant cette stratégie, nous avons identifié trois voies dominantes montrant l'activation dans la majorité (> 70%) de GCS: prolifération /cellules souches, Wnt /β-caténine, et la signalisation NF-kB La capacité à effectuer. tels "à haut débit voie profilage» ouvre de nombreuses pistes intéressantes. Par exemple, plusieurs études ont déjà rapporté des résultats contradictoires concernant l'impact pronostique de différentes voies oncogéniques en GC - les implications pronostiques des antigènes liés à la prolifération tels que Ki-67 en GC ne sont pas fermement établies [29], et l'activation élevée NF-kB GC a été associée à la fois bon et mauvais résultat GC patient dans différentes études [7], [30]. Il est tout à fait possible que certains de ces incohérences peuvent avoir été en raison d'une mise au point historique sur l'utilisation des méthodes classiques et l'analyse soit des voies simples ou individuelles composants de la voie (gènes /protéines). Notre observation que les combinaisons de la voie sont prédictifs des résultats pour les patients suggère que les combinaisons de la voie, plutôt que seuls les voies simples, peuvent jouer un rôle crucial pour influencer le comportement de la tumeur. Un autre avantage de haut débit voie profilage est la capacité à définir supérieur des relations d'ordre entre les voies oncogéniques distinctes. Dans l'étude actuelle, nous avons constamment observé une activation concomitante de E2F, MYC, p21 (-repression), et la tige des voies cellulaires dans les tumeurs (le «/cellules souches prolifération" cluster de voie). Ceci est probablement dû à une augmentation de la prolifération cellulaire dans les cellules tumorales, comme E2F est important dans le contrôle de la prolifération cellulaire et MYC est à la fois p21 répresseur et inducteur de la cycline D2, cycline-dépendante et la liaison protéine-kinase CksHs2 [31]. En outre, les cellules souches, en particulier des cellules souches embryonnaires (CSE), sont également connus pour présenter des taux élevés de prolifération cellulaire [32]. Plus curieusement, nous avons également observé des associations étroites entre apparemment fonctionnellement différentes voies, comme β-caténine et SRC, ainsi que l'inhibition des HDAC et BRCA1. De tels motifs voie co-activation peuvent suggérer des interactions fonctionnelles entre ces voies, qui méritent d'être étudiés plus loin. Par exemple, il est possible que l'activation de c-SRC peut augmenter l'expression de la signalisation de voie Wnt [33]. Explorer les relations entre les voies montrant co-activation peut ainsi fournir des informations précieuses quant à la capacité de la cellule cancéreuse pour coordonner l'activité des voies multiples. Un troisième avantage de l'approche voie de profilage est qu'il facilite l'identification des principaux voies liées à la maladie. Parmi les voies analysées dans cette étude, la constatation que la signalisation NF-kB peut être élevée dans une proportion importante de GCS mérite une certaine attention que cette voie a été relativement moins explorée dans GC. Fait intéressant, alors que nous avons observé une différence significative dans les deux p50 et p65 (les sous-unités NF-kB) l'expression génique entre /off GCCLs NF-kB /sur et NF-kB, nous n'avons pas observé manifeste expression de la protéine p50 différentielle dans ces lignes, en Contrairement à p65 (figure 4C). Cela peut être dû à une combinaison de trois raisons. Tout d'abord, la gamme absolue de l'expression du gène p65 dans les lignées cellulaires est nettement supérieure à la plage absolue de l'expression du gène p50 (> 3 ×, Figure S4). D'autre part, l'essai par Western blot utilisée pour effectuer ces mesures de protéine est connue pour être hautement non quantitative, ce qui peut masquer les différences subtiles dans l'expression. En troisième lieu, au-delà de l'expression des gènes, l'expression de p50 est également soumis à une variété de mécanismes de régulation post-transcriptionnelle tels que le précurseur de clivage qui pourrait affecter le niveau final de la protéine p50, tandis que p65 est pas généré à partir d'une protéine précurseur [34]. NF-kB a été montré pour être activé par H. pylori le in silico En conclusion, nous avons montré dans ce travail que les voies signatures peuvent être utilisées avec succès pour prédire l'état d'activation des voies de signalisation cellulaire, même dans des entités biologiques aussi complexes en tant que CG humaine. Une application immédiate évidente de ces taxonomies basé voie-peut se rapporter à l'utilisation des thérapies ciblées. Les premiers essais évaluant le rôle des thérapies ciblées en GC ont démontré que des résultats modestes [38]; cependant, la plupart de ces études ont été réalisées sans que les patients pré-stratifier en utilisant des critères moléculaires ou histopathologiques.
stratégie pour cartographier les modèles d'activation de la voie oncogénique dans 301 cancers gastriques primaires, la deuxième cause de mortalité dans le monde du cancer. Nous avons identifié trois voies oncogéniques (prolifération /souches cellulaires, NF-kB, et Wnt /β-caténine) dérégulé dans la majorité (> 70%) des cancers gastriques. Nous avons validé fonctionnellement ces prédictions de la voie dans un panel de lignées cellulaires de cancer gastrique. stratification des patients par des combinaisons de la voie oncogènes a montré des différences de survie reproductibles et significatives dans plusieurs cohortes, ce qui suggère que les interactions de la voie peuvent jouer un rôle important pour influencer le comportement de la maladie. GCS individuels peuvent être taxonomized avec succès par activité de la voie oncogénique dans biologiquement et cliniquement sous-groupes pertinents. Prédiction de l'activité de la voie par des signatures d'expression permet ainsi l'étude des voies multiples liés au cancer qui interagissent simultanément dans les cancers primaires, à l'échelle actuellement pas réalisable par d'autres plates-formes.
Auteur Résumé
méthode pour cartographier les niveaux de différentes voies d'activation dans les cohortes de profils de tumeurs primaires complexes et cette approche de classification de la voie dirigée en utilisant des exemples de preuve de concept d'un cancer du sein validé. Nous avons ensuite appliqué cette méthode pour GC pour évaluer les voies oncogéniques onze précédemment impliqués dans la carcinogenèse gastrique [3] - [7], [12] - [17]. Au total, nous avons analysé plus de 300 GCS primaires dérivées de trois cohortes indépendantes de patients, d'effectuer au mieux de notre connaissance la plus grande analyse génomique de GC à ce jour. Nous avons identifié trois voies oncogéniques (facteur-kB nucléaire (NF-kB), Wnt /β-caténine, et la prolifération /cellules souches) qui ont été déréglementé dans la grande majorité (> 70%) de glucocorticoïdes, et fonctionnellement validé les prédictions de la voie in vitro
utilisant un panel de lignées cellulaires de GC. Bien que la stratification des patients au niveau des parcours individuels n'a pas réussi à démontrer constamment des différences significatives dans les résultats cliniques, la stratification des patients par des combinaisons de la voie oncogéniques (par exemple forte prolifération /haute NF-kB par rapport à faible prolifération /faible NF-kB) a montré des différences de survie reproductibles et significatives dans plusieurs cohortes indépendantes de patients, ce qui suggère un rôle critique pour les combinaisons de la voie pour influencer le comportement clinique de GC. Nos résultats démontrent donc que GCS peuvent être taxonomized avec succès en utilisant activité de la voie oncogénique dans biologiquement, fonctionnellement et cliniquement pertinente des sous-types.
les de
ou in vivo
système expérimental bien défini. En second lieu, nous avons cartographié les signatures de la voie sur les profilés d'expression génique à partir d'une série hétérogène de cancers. En troisième lieu, selon une procédure non paramétrique pattern matching basé rang, les scores d'activation ont été affectés à des cancers individuels basés sur la force de l'association à la signature de la voie. Enfin, les cancers individuels ont été triés en fonction de leurs scores d'activation de la voie.
stratégie dans une série de preuve de expériences -principle. Nous avons choisi l'exemple du cancer du sein, une tumeur maligne pour laquelle il existe de nombreuses preuves de l'hétérogénéité et de la voie «sous-types moléculaires" discrets [18]. Pour effectuer cette validation, nous avons d'abord demandé si les signatures de voie précédemment décrits sont associés à la signalisation des oestrogènes altérée peuvent être utilisées pour identifier des lignées de cellules de cancer du sein présentant des niveaux élevés de récepteur d'oestrogènes (ER) d'activité. Nous avons analysé un panneau d'expression génique des lignées cellulaires de cancer du sein 51 décrits initialement dans Neve et al. [18] avec un 11 gène «tamoxifène sensibilité» de la signature de la voie dérivée d'une liste de gènes exprimés de manière différentielle entre MACA 3366, un mammaire humain carcinome xénogreffe tamoxifène sensible, et maca 3366 /TAM, une sous-lignée de tamoxifène résistant du même xénogreffe [19]. Nous avons constaté que les lignées cellulaires de cancer du sein positivement associé à la signature de la sensibilité du tamoxifène présentait des taux d'expression significativement plus élevé de ESR1
, le récepteur de l'oestrogène et de la cible moléculaire de tamoxifène, par rapport aux lignes montrant négatives scores d'activation de la voie (p = 2,12 × 10 -7, Exactitude 84,3%, Sensibilité 100%, Spécificité 75%) (figure 1B et le tableau S1).
Patterns de oncogénique activation de la voie dans le GC
validation de
Pathway Prédictions /cellules souches) qui ont été activés individuellement dans une proportion significative des glucocorticoïdes (≥35%), et lorsqu'il est combiné couverture de la majorité (>fourni; 70%) de GCS. Prolifération /souches voies cellulaires ont été activées dans 40% des glucocorticoïdes dans chaque cohorte (plage: 38 à 43%), les voies Wnt /β-caténine ont été activés dans 46% des glucocorticoïdes (plage: 43 à 48%), et le NF- voie kB a été activé dans 39% des glucocorticoïdes (plage: 35 à 41%) (barres de couleur en dessous de chaque heatmap dans la figure 2). Ces fréquences et d'autres voies souvent déréglementés (par exemple p53) sont répertoriés dans le tableau S3.
prédictions de la voie en utilisant les profils d'expression des gènes sont associés à l'activation de la voie pertinente in vitro
.
Discussion
[35], un cancérogène connu GC et aberrante de signalisation NF-kB a également été impliqué dans plusieurs cancers inflammatoires liés tels que GC [36]. NF-kB a été suggéré d'être activée constitutivement dans les cancers gastriques primaires dans quelques études [7]. NF-kB-inhibiteurs ciblés sont actuellement développés activement dans de nombreux programmes de développement de médicaments anticancéreux et d'un sous-ensemble de patients du GC (ceux avec l'activité de NF-kB élevée) peuvent représenter une sous-classe appropriée pour évaluer l'efficacité de ces composés.
méthode utilisée dans notre étude est conceptuellement similaire aux travaux de Bild et al, qui a utilisé un modèle de régression binaire pour classer les tumeurs en fonction de l'activité prévue de cinq voies oncogéniques [9]. Contrairement à la régression binaire, notre approche, ce qui rend l'utilisation d'une connectivité métrique à base de rang [37], ne nécessite aucun processus de formation élaboré sur chaque signature de la voie et ne nécessite pas la disponibilité des données d'expression premières, ce qui facilite l'utilisation des nombreux public des signatures de la voie dans la littérature [27]. Cependant, l'approche basée sur l'expression du gène a des limites. Tout d'abord, parce que nos prédictions de la voie sont basées sur l'expression des gènes plutôt que des protéines, telles prédictions sont des substituts certes moléculaires de la véritable activité de signalisation de voie. Deuxièmement, nous sommes actuellement limités à l'analyse connue voies oncogéniques précédemment identifiées dans la littérature. Troisièmement, bien que nous avons pu utiliser des signatures de la voie à partir des contextes très différents de tissus pour prédire l'état d'activation de la voie, un examen des premiers exemples de preuve de principe cancer du sein a révélé que l'association du statut de ER à l'oestrogène réactivité comme prédit en utilisant la signature de l'ostéosarcome , bien qu'important, est nettement plus faible par rapport à l'association de l'état d'urgence à la sensibilité tamoxifène prédite en utilisant une signature dérivée du même type de tissu (par exemple du sein). Ce résultat implique que il peut aussi exister des différences spécifiques de tissus dans la voie des signatures qui peuvent affecter la précision de la prédiction. Quatrièmement, par rapport à notre étude qui a porté sur les voies de pertinence biologique connue en GC, on ne sait pas si cette méthode peut être appliquée à des maladies où la connaissance préalable des voies concernées peuvent ne pas être disponibles. Toutefois, il convient de noter qu'une multitude de signatures de la voie (> 1000) associée à diverses voies biochimiques et de signalisation existe déjà dans la littérature, qui peuvent être accessibles à partir des bases de données publiques telles que MSigDB (http://www.broad.mit .edu /GSEA /msigdb /genesets.jsp? collection = CGP). Depuis notre approche peut être appliquée à pratiquement tout ensemble de données de la maladie pour laquelle l'information de l'expression des gènes est disponible, tester chaque signature d'une manière à haut débit pour des preuves de la voie de déréglementation est à la fois concevable et réalisable. Dans de tels cas, la voie présentant des hautes fréquences de la déréglementation représenterait alors les voies de candidats impliqués dans la maladie en question, qui peuvent ensuite être ciblées pour la recherche ciblée et l'expérimentation. Aborder ces questions constitueront le terrain pour beaucoup de recherches futures
.
 Faire face au syndrome du côlon irritable
Faire face au syndrome du côlon irritable
 Les bactéries intestinales peuvent prédire le risque d'hypertension pulmonaire
Les bactéries intestinales peuvent prédire le risque d'hypertension pulmonaire
 Le sexe oral peut déclencher une vaginose bactérienne
Le sexe oral peut déclencher une vaginose bactérienne
 Des microbes sur la langue pourraient être utilisés pour diagnostiquer le cancer du pancréas
Des microbes sur la langue pourraient être utilisés pour diagnostiquer le cancer du pancréas
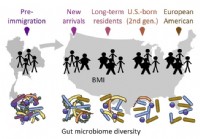 La migration affecte le microbiote intestinal qui à son tour affecte la santé
La migration affecte le microbiote intestinal qui à son tour affecte la santé
 Un champignon commun trouvé sur la peau peut provoquer une maladie inflammatoire de l'intestin
Un champignon commun trouvé sur la peau peut provoquer une maladie inflammatoire de l'intestin
 Le pH acide améliore l'infection par le SRAS-CoV-2 en régulant à la hausse le récepteur ACE2
La pandémie actuelle de la maladie à coronavirus 2019 (COVID-19) qui est causée par un nouveau coronavirus, à savoir, syndrome respiratoire aigu sévère coronavirus 2 (SARS-CoV-2), a fait plus de 4,6 m
Le pH acide améliore l'infection par le SRAS-CoV-2 en régulant à la hausse le récepteur ACE2
La pandémie actuelle de la maladie à coronavirus 2019 (COVID-19) qui est causée par un nouveau coronavirus, à savoir, syndrome respiratoire aigu sévère coronavirus 2 (SARS-CoV-2), a fait plus de 4,6 m
 Un régime végétalien pourrait stimuler les microbes intestinaux qui aident à la perte de poids
Passer à un régime végétalien pourrait aider les gens à perdre près dune livre de poids chaque semaine et réduire considérablement leur risque de diabète, selon une étude récemment présentée lors de l
Un régime végétalien pourrait stimuler les microbes intestinaux qui aident à la perte de poids
Passer à un régime végétalien pourrait aider les gens à perdre près dune livre de poids chaque semaine et réduire considérablement leur risque de diabète, selon une étude récemment présentée lors de l
 Le microbiote intestinal pourrait prédire la gravité du COVID-19
La pandémie de COVID-19 se propage aux quatre coins du monde. Mais tout le monde ne tombe pas malade au même rythme. Une nouvelle étude publiée sur le serveur de préimpression medRxiv en avril 2020
Le microbiote intestinal pourrait prédire la gravité du COVID-19
La pandémie de COVID-19 se propage aux quatre coins du monde. Mais tout le monde ne tombe pas malade au même rythme. Une nouvelle étude publiée sur le serveur de préimpression medRxiv en avril 2020