Abstrakt
Upptäckten av effektiva anti-cancer läkemedelskombinationer är en stor utmaning, eftersom experimentell testning av alla möjliga kombinationer är uppenbart omöjligt. Nya insatser för att beräkningsförutsäga läkemedelskombination svar behålla detta experimentella sökrymden, som modelldefinitioner grundar sig oftast på omfattande drogstörningsdata. Vi utvecklade en dynamisk modell som representerar en cell öde beslut nätverk i AGS magcancer cellinje förlita sig på bakgrundskunskap utvinns ur litteratur och databaser. Vi definierade en uppsättning av logiska ekvationer rekapitulera AGS data som observerats i celler i deras baseline proliferativa tillstånd. Med användning av modelleringsmjukvara GINsim, reduktion modellen och simulering kompressionstekniker tillämpades för att klara av den stora tillståndsrymden av stora logiska modeller och möjliggöra simuleringar av parvisa tillämpningar av specifika signalerings inhibitoriska kemiska ämnen. Våra simuleringar förutspådde synergistisk tillväxthämmande verkan av fem kombinationer av totalt 21 möjliga par. Fyra av de förväntade synergierna bekräftades i AGS celltillväxt realtidsanalyser, inklusive kända effekter av kombinerad MEK-AKT eller MEK-PI3K hämningar, tillsammans med nya synergieffekter av kombinerad TAK1-AKT eller TAK1-PI3K hämningar. Vår strategi minskar beroendet av a priori drog störnings experiment för väl karakteriserade signalledningar, genom att visa att en modell förutsäga kombiläkemedelseffekter kan härledas från bakgrunds kunskap om ostörda och förökar cancerceller. Vår modellering kan därmed bidra till preklinisk upptäckten av effektiva läkemedel mot cancer kombinationer, och därmed utvecklingen av strategier för att skräddarsy behandlingen för enskilda cancerpatienter.
Fighting cancer med kombinationer av läkemedel ökar framgången för behandling. Men på grund av det stora antal läkemedel och tumörvarianter, är det fortfarande en enorm utmaning att identifiera effektiva kombinationer. För att illustrera detta, en uppsättning av 150 läkemedel motsvarar mer än 10.000 möjliga parvisa kombinationer av läkemedel. Experimentell testning av alla möjligheter är uppenbart omöjligt. Vi har utvecklat en beräkningsmodell som tillåter oss att identifiera förmodligen effektiva kombinationer, och som samtidigt visar kombinationer som kan vara utan effekt. Modellen är baserad på specifika cancercell biomarkörer som erhållits från ostörda cancerceller, och används sedan för att utföra omfattande automatiserad logiskt resonemang. Laboratorietester av läkemedelssvar förutsägelser bekräftade resultat för 20 av 21 läkemedelskombinationer, däribland fyra av fem läkemedels par förutspådde att synergistiskt hämma tillväxten. Vår strategi är relevant för preklinisk upptäckten av effektiva läkemedel mot cancer kombinationer, och därmed för att utveckla strategier för att skräddarsy behandlingen för enskilda cancerpatienter
Citation. Flobak Å, Baudot A, Remy E, Thommesen L, Thieffry D , Kuiper M, et al. (2015) Upptäckt av drog Synergier i Gastric cancerceller förutsägs av Logical Modeling. PLoS Comput Biol 11 (8): e1004426. doi: 10.1371 /journal.pcbi.1004426
Redaktör: Ioannis Xenarios, schweiziska institutet för bioinformatik, USA
emottagen: 5 mars 2015; Accepteras: 3 juli 2015, Publicerad: 28 augusti 2015
Copyright: © 2015 Flobak et al. Detta är en öppen tillgång artikel distribueras enligt villkoren i Creative Commons Attribution License, som tillåter obegränsad användning, distribution och reproduktion i alla medier, förutsatt den ursprungliga författaren och källan kredit
datatillgänglighet: Alla relevanta uppgifter är inom pappers- och dess stödjande information filer
Finansiering:. Detta arbete stöddes av Liaison Committee mellan det centrala Norge Regional Health Authority (RHA) och Norges teknisk-naturvetenskapliga universitet (NTNU). Ett besök finansierades av EMBO Short Stay gemenskap för AB till NTNU, Norge, oktober-november 2013. finansiärerna hade ingen roll i studiedesign, datainsamling och analys, beslut att publicera, eller beredning av manuskriptet.
konkurrerande intressen:. författarna har deklarerat att inga konkurrerande intressen finns
Introduktion
Det har länge varit tänkt att framtida anticancerbehandling kommer att anta kombinatoriska metoder, där flera specifika anti-cancerläkemedel tillsammans rikta flera robusthets funktioner eller svagheter i en särskild tumör [1-3]. Effektiviteten av kombinatoriska anti-cancerbehandlingar kan ytterligare maximeras genom att utnyttja synergiläkemedels åtgärder, vilket innebär att olika läkemedel administreras tillsammans uppvisar en förstärkte effekt jämfört med de enskilda läkemedel. Drog synergi är attraktiv eftersom den möjliggör en signifikant reduktion i doseringen av de enskilda läkemedlen, men behåller den önskade effekten. Synergier håller därför potential att öka behandlingseffektivitet utan att trycka enstaka läkemedelsdoser till nivåer där de leder till biverkningar. Därför synergier som identifierats i prekliniska studier utgör intressanta kandidater för ytterligare karakterisering i cancermodeller och kliniska prövningar.
Det pågående arbetet för att identifiera positiva kombinatoriska anti-cancerterapier typiskt förlitar sig på storskaliga experimentella störningsdata antingen för att besluta om specifik patientbehandling [4], eller för prekliniska pipelines att föreslå nya läkemedelskombinationer [5-8]. Detta arbete står emellertid inför utmaningar som den stora sökrymden som måste stödjas av experimentella data, vilket gör systematiska sökningar för effektiva kombinationer utmanande. Dessutom antal villkor för provning dramatiskt ökar när man överväger högre ordningens kombinationer, flertal läkemedelsdoser, temporal optimering av läkemedelsadministration, och mångfald typer av cancerceller och patienter. Således måste lösningar sökas för att minska den experimentella sökrymden av läkemedelskombinationer och deras programlägen för att få en kvalificerad repertoar av kombinationsterapier för kliniska prövningar, och slutligen för att stödja leverans av personlig service.
Computational modeller används alltmer för att förutsäga läkemedelseffekter [6,9], i syfte att rationalisera och hushålla den experimentella flaskhals. För att möjliggöra väsentlig reduktion av antalet relevanta villkor som måste testas, skulle sådana modeller idealt konstrueras utan behovet av massiva experimentella drogen stördata. Strategier där utformningen av prediktiva modeller kan baseras på molekylära data från ostörda cancerceller är därför attraktiva.
Vi bestämde oss för att fokusera på Boolean och multilevel logiska modeller, eftersom de möjliggör en relativt enkel formalisering av orsakssammanhang inbäddade i molekylära nätverk, såsom signalöverföring och regulatoriskt gennätverk. Dessutom kan logiska modellsimuleringar användas för att automatisera resonemang på nätverksdynamik, även med knappa kunskaper om kinetiska parametrar [10-15], och har använts för att beskriva och förutsäga beteendet hos molekylära nätverk som berörs i mänskliga sjukdomar [13,14] . Sådana modellerings ansträngningar har bidragit till förståelsen av mekanismerna bakom tillväxtfaktor inducerade signalering i cancerceller och valet av kandidat målprotein för nya anticancerbehandling [16-23]. Medan tidigare studier har visat kraften av logiska modeller för att förutse enda läkemedel åtgärder, sträcker vi användning av logiska modellering för att förutsäga effekterna av kombinato hämning av två eller flera signalöverföringskomponenter.
Vi rapporterar konstruktionen av en logisk modell som omfattar molekylära mekanismer som är centrala för att kontrollera celltillväxt av adenocarcinom cellinje AGS. Efter en inledande montering av en omfattande signalering och reglerande nätverk från allmän signaltransduktion kunskap, de logiska regler som är förknippade med var och en av de 75 modellkomponenter förfinas med hjälp av grundläggande data som erhållits från aktivt växande AGS celler. Den resulterande logisk modell användes för att bedöma drogsynergipotentialen bland 21 parvisa kombinationer av sju kemiska hämmare, var inriktad på ett specifikt signalerande komponent. Modellsimuleringar föreslagit fem kombinationer av inhibitorer att vara synergistisk, av vilka fyra kunde senare bekräftas i celltillväxt experiment. Viktigt är ingen av kombinationerna förutspåtts av modellen att vara icke-synergistisk visade synergistiska tillväxthämmande effekter i våra cellulära analyser, var dvs inga falska negativa observerats. Våra resultat visar att vår logisk modell, konstruerad utan användning av initiala storskaliga hämmare störningsdata rekapitulerar viktiga molekylära reglerande mekanismerna bakom tillväxten av AGS-celler på ett sätt som gör det möjligt framgångsrik förutsägelse av den synergistiska effekten av hämmare kombinationer i experimentella cellkulturer. Med ledning av den modell, identifierade vi två etablerade synergistiska läkemedelsinteraktioner och upptäckte två synergier inte tidigare rapporterats.
övergripande strategi för att förutsäga och validering av läkemedels synergier
för att upptäcka kombiläkemedelsbehandlingar synergistiskt utövar hämning av cancercellernas tillväxt, har vi utvecklat ett arbetsflöde som kombinerar numeriska och experimentella analyser för att förutsäga och utvärdera läkemedels synergier (Fig 1).
Våra modellering förfarandet integreras a priori Efter en modell reduktionssteg, där noder och logiker som hänför sig till nya läkemedelsmål och fenotypiska utgångar bibehålls, modellen används för uttömmande simuleringar av effekterna av parvisa nod hämningar med sju kända kemiska hämmare. Slutligen tillväxthämmande effekter av dessa läkemedelskombinationer på AGS celler testas experimentellt. Konstruktion av en reglerande graf som omfattar viktiga signalvägar. AGS celler hamnen mutationer i många gener som kodar viktiga signalsystem komponenter som är kända för att avregleras i magcancer till exempel komponenter av MAPK, PI3K, Wnt /β-catenin och NF-kB vägar [24,25]. Baserat på erfarenheter från databaser och vetenskapliga publikationer, har vi integrerat information om MAPK vägar (JNK, p38 MAPK och ERK), de PI3K /AKT /mTOR vägar, Wnt /β-catenin vägen, och NF-kB-vägen, såväl som överhörning mellan dessa vägar (se fig 2, Material och Metoder och S1 Text). Den resulterande nätverket består av 75 signalering och regulatoriska komponenter (proteiner, proteinkomplex och gener) och 149 riktade interaktioner. Två avläsning noder (utgångar), som heter Prosurvival Mössor och Antisurvival Reglerings nätverket omvandlades till en logisk modell, där den lokala aktivitet tillståndet för varje komponent (nod) representerades av en boolesk variabel (med värdena 0 eller 1). Några noder var förknippade med flera nivåer variabler: de två utgångsnoder, Prosurvival Mössor och Antisurvival När som helst, är den globala systemets tillstånd representeras av en diskret vektor innehållande Boolean eller fleraktivitetsvärden för alla nätverkskomponenter [26]. Eftersom alla noder stater iterativt uppdateras i simuleringar modellen konvergerar till sina attraktorer, företrädd av enstaka globala fasta stater i enkla attraktorer, eller uppsättningar av stater upprepade gånger passeras i komplexa attraktorer. Baserat på den rättsliga grafen och definierade logiska regler ovan använde vi en kraftfull algoritm implementeras i GINsim att beräkna alla stabila tillstånd av modellen. För att kalibrera modellen med avseende på aktivt växande AGS celler, jämförde vi nod tillstånd förutsägelser mot AGS baslinjen biomarkörer observationer rapporterats i litteraturen. Vi har granskat 72 vetenskapliga publikationer och fann 219 experiment med prolifererande AGS-celler med information om aktiviteten hos proteiner som representeras i vår modell (se S1 Text och S2 tabell). Vi valde en delmängd av 21 proteiner för vilka aktivitetsdata stöddes av flera oberoende men konsekventa rapporter. Med hjälp av dessa experimentella observationer som riktlinjer för "gold standard" proteinaktiviteter i aktivt växande AGS-celler, vi jämförde tillståndet för var och en av dem med deras nivå i den beräknade attractor av modellen. För att få en enda stabilt tillstånd innehållande aktivitetsnivåer i alla modellkomponenter, har de logiska regler för de delar av ERK-vägen (SHC1, SOS, Raf, MEK och ERK) definieras för att återspegla den observationen att ERK är aktiv i förökande AGS-celler (se S1 Text och S2 tabell). Efter dessa ändringar, visade modellen optimeras: den observerade attractor av ostörda modellen var ett stabilt tillstånd noggrant bekräftas av experimentella observationer i ostörda växande AGS celler, som värdena för alla 21 noder som vi kunde kontrollera matchen rapporterade protein aktiviteter (se S1 Text, S3 och S4 tabeller). Dessutom värdet av avläsnings noderna Prosurvival för att bedöma kombinationer av hämningar för samverkan har vi fokuserat på den systematiska hämning av sju modell noder och deras 21 parvisa kombinationer. Dessa sju noder (märkta med tjocka kanter i fig 2) valdes eftersom potenta och specifika kemiska hämmare fanns tillgängliga för inriktning motsvarande proteinkinaser i biologiska experiment (tabell 1). Använda en asynkron uppdatering politik (se Material och metoder), simulerade vi effekten av kemiska hämningar genom att tvinga staten riktade modell noder vara 0 (inaktiv), och sedan beräkna den resulterande attraktor. Varje hämning av enskilda noder eller par av noder ledde till en unik attraktor. I ett fåtal fall nådde systemet en komplex attraktor, där en delmängd av stater förs upprepade gånger (se Material och Metoder och S1 text). Beräkningen av potentiella komplexa attraktorer är utmanande på grund av den kombinatoriska explosion av stater för stora logiska modeller. För att klara av detta problem använde vi en modell minskning metod för att erhålla en komprimerad modell bevara de utvalda läkemedelsmål och kompakttillståndsövergången diagram i ett hierarkiskt sätt (se Material och Metoder och [14]). Den reducerade logisk modell (se fig 3 och S3 dataset) erhölls genom att iterativt ta bort komponenter inte riktade vid droger, och var tillräckligt liten för att möjliggöra uttömmande asynkrona simuleringar och noggrann karakterisering av både stabila tillstånd och komplexa attractors, vilket därigenom möjliggör analys av all enda och par av hämningar. för att underlätta tolkningen, definierade vi den totala svaret tillväxt tillväxt (perturbation1 & perturbation2) < Min (tillväxt (perturbation1), tillväxt (perturbation2)), Till exempel, tillväxt (perturbationMEK & perturbationAKT) Review = 0,5; vilket är ett värde som är lägre än vad som observerats med störningar av antingen MEK eller AKT: tillväxt (perturbationMEK) Review = 1,5; tillväxt (perturbationAKT) Review = 2. Simuleringarna förutspådde fem synergistiska kombinationer (< 25% av de 21 möjliga par). Tre av dessa kombinationer involvera MEK, tillsammans med PI3K, AKT eller p38. De två återstående synergier innebär TAK1 med antingen PI3K eller AKT (Fig 4). För att bedöma giltigheten av våra modellprediktioner, en cellanalys i realtid användes för att testa kemiska inhibitorer av de sju proteiner (Tabell 1) för sin förmåga att begränsa AGS celltillväxt i enkel- och kombinatoriska formuleringar. effekten av kemiska inhibitorer analyserades med användning av en strategi baserad på Loewe: s definition av synergi [28 ], där det anges att en synergistisk interaktion presterar bättre än den förväntade additiva effekten observerades när en inhibitor kombineras med sig i en "noll-interaktion" experiment. För att kvantifiera synergistiska interaktioner, var en kombinatorisk index (CI) beräknad [29], baserad på tillväxt mätt 48 timmar efter tillsats av inhibitorer. CI-värden sträcker sig från noll till oändligheten, och värden under 1 indikerar synergistiska interaktioner. Fyra av de fem synergier förutsagts av vår logisk modell bekräftades experimentellt, med CI värden långt under 0,5, vilket indikerar en stark synergi. I själva verket var en djupgående effekt på AGS celltillväxt hittades när MEK eller TAK1 hämmare kombinerades med PI3K eller AKT-hämmare. Motsvarande tillväxtkurvor (Fig 5) visar att celltillväxt i närvaro av två hämmare kombineras vid halv sina GI50 koncentrationer (lila kurvor) är varje betydligt lägre än tillväxten i närvaro av antingen inhibitor ensam vid sin fulla GI50 koncentration (grönt och blå kurvor). Däremot kan en kombination av MEK och p38-hämning inte bekräftas i celltillväxtexperiment. Viktigt, observerade vi inga falska negativa prognoser, vilket innebär att den återstående hämmare kombinationer, förutspådde att sakna synergieffekter, verkligen misslyckats med att visa synergi i våra cellulära analyser. Sammantaget våra modellsimuleringar har visat sig vara mycket exakt, korrekt förutsäga effekterna av 20 av de 21 kombinationer. Synergieffekter av PI3K-MEK eller AKT-MEK hämningar har redan setts i en mängd olika tumörceller [ ,,,0],6,30-34], vilket ger ytterligare förtroende för synergierna i TAK1-PI3K och TAK1-AKT hämningar. Därför är dessa nya kombi hämningar lovande kandidater lätt mottagliga för experimentell testning i en rad olika typer av cancerceller. Att förstå signalerings mekanismerna bakom synergistiska hämningar är av stort intresse eftersom det kan bidra till att identifiera biomarkörer informativa av behandlingssvar, som kan tjäna som guider att välja mellan en arsenal av etablerade läkemedels synergier rätt behandling för den enskilda patienten. Undersökning av simulerade störningseffekter med vår AGS logisk modell visade att FOXO, representerande pro-apoptotiska transkriptionsfaktorer inaktiveras av fosforylering [35], var synergistiskt aktiveras av kombinerad MEK och PI3K eller MEK och AKT inhibition (se S1 text). Intressant nog enda hämmande störning av MEK, PI3K eller AKT inte ändra FOXO aktivitet (se S1 text för mer information). Dessa observationer stämmer experimentella resultaten i humana umbilikalvensendotelceller (HUVEC), där inhibitorer inriktade MEK och AKT rapporteras till synergistiskt aktivera FOXO [36], vilket tyder på att våra modellsimuleringar kan utgöra en grund för biologiskt relevanta hypoteser om molekylära effekter nedströms specifika hämmare. för att ytterligare undersöka mekanismerna involverar FOXO vi simulerade störningarna inhibitor i en FOXO knock-out-modell, och fann att kombinerad hämning av MEK och PI3K visade ingen ökad tillväxt hämmande effekt jämfört med deras motsvarande enda hämning . Detta tyder på att MEK-PI3K synergi beror faktiskt på FOXO. För den kombinerade MEK och AKT inhibition, de FOXO knock-out modellsimuleringar visade endast en mindre minskning av den synergistiska effekten av kombinerad MEK och AKT inhibition. Synergin mellan MEK och AKT-hämmare förefaller således vara mindre beroende av FOXO. Sammantaget dessa simulerings resultat tyder potentiellt intressanta skillnader mellan pro-apoptotiska signaleringshändelser när man jämför MEK-AKT inhibering mot MEK-PI3K inhibition. Den mekanistiska grund av de synergier som observerats när hämma TAK1-AKT, eller TAK1- PI3K, är okänd. Intressant, AGS modellsimuleringar visar att FOXO aktiveras när TAK1 inhiberas i kombination med antingen PI3K eller AKT, men inte av enskilda hämningar. Aktivering av FOXO är således en potentiell medlare även för synergier som involverar TAK1. Till stöd för detta, simuleringar visade att både TAK1-PI3K och TAK1-AKT synergier avskaffades när FOXO slås ut, i likhet med konstaterandet av MEK-PI3K hämning (se S1 text). Potentiellt, ERK kunde vara inblandad i signalering nedströms TAK1. I det fallet MAP kinaskaskad kan utgöra en gemensam mekanism inblandad i de synergier som involverar MEK-PI3K och MEK-AKT, och de involverar TAK1-PI3K och TAK1-AKT. Men våra AGS modellsimuleringar förutspår att ERK är fortfarande aktiv efter kombinerad hämning av TAK1 och PI3K eller TAK1 och AKT. Detta kan tyda på att MEK /ERK inte är involverad i de efterföljande effekterna av de hämmande störningar som involverar TAK1. En annan kinas skulle kunna fungera som en punkt av överhörning för TAK1 och PI3K /AKT-signalering. I detta avseende NLK (Nemo-liknande kinas) är en intressant kandidat eftersom det är känt att agera nedströms TAK1 [37], medla hämmande fosforylering av FOXO [38]. modellbaserad förslag att FOXO aktivering kan vara viktig för synergistisk tillväxthämning inte hitta experimentellt stöd i flera konton FOXO proteiner som fungerar som förmedlare av cytotoxiska kemoterapeutiska läkemedel [39]. Detta tyder på att den dynamiska beteende vår logisk modell rekapitulerar generiska egenskaper som kan vara relevanta för en rad olika tumörtyper. Utvecklingen av nya anti-cancer medicin fokuserar främst på droger riktade mot specifika molekylära mål. Emellertid har kliniska tillämpningar ofta varit en besvikelse, vilket endast övergående svar följt av läkemedelsresistens som hindrar terapifördelar. Detta har lett till att behandlingen av terapier baserade på kombinationer av läkemedel inriktade på olika signalvägar eller cellulära processer, i syfte att hindra utvecklingen av läkemedelsresistens och samtidigt möjliggöra en minskning av läkemedelsdosering, att sänka läkemedelsinducerad toxiska effekter [2,3,40,41]. Dessa starka incitament för kombinatorisk läkemedelsbehandling utmanas av de många kombinationer för att ta hänsyn till och genom det faktum att effekten av en given läkemedelskombination är beroende av arten av den specifika tumören. Således, för att upptäcka apt läkemedelskombinationer i en takt kompatibel med den stora sökrymden utgörs av de många läkemedel och olika cancercell spektrum, är det obligatoriskt att utveckla effektiva strategier för att förutsäga gynnsam kombinationsbehandling för enskilda cancerformer. Aktuell ansträngningar för att komma till ett rationellt val av läkemedelskombinationsbehandling med hjälp av primära tumörcellkulturer och xenograftstudier konfronteras med höga kostnader och en rörlig ränta på framgång i tumörcelltillväxthämning, och kämpar för att erhålla mycket exakta förutsägelser inom den tidsram som begränsas av sjukdom progression [4,42-44]. Medan cancer cellinjekulturer sällan tillåter upptäckter som direkt kan överföras till en klinisk miljö, de möjliggör experimentella undersökningar av mekanismerna bakom den biologiska mångfalden och robusthet och kan således användas för att undersöka strategier för att identifiera potentiellt effektiva läkemedel kombinationsbehandlingar. De kan därför bidra till att skapa en stor arsenal av fördelaktiga kombinationer av läkemedel som åtföljs av prognosverktyg som gör det möjligt att välja rätt kombination för den enskilde tumören. Men även i dessa cellulära modeller, är det inte möjligt att testa alla potentiella läkemedelskombinationer och programlägen för en tillräcklig spektrum av typer av cancerceller. I detta sammanhang kan datormodellering vara till stor hjälp för att minska den experimentella sökrymden. Vi har visat hur en logisk modell byggs från känd information signaltransduktion nätverk kan skräddarsys till en specifik cancercell-system med användning av utgångsdata , så att den kan användas för att förutsäga synergistiska och icke-synergistiska kombinatoriska tillväxthämmande behandlingar. Fyra av de fem förväntade synergistiska kombinationer bekräftades experimentellt med några falska negativa prognoser. Med en sådan framgång, skulle det ha varit tillräckligt för att testa endast en fjärdedel av de 21 möjliga kombinationer av läkemedel som undersökts och fortfarande inte missa någon synergistisk par. Våra resultat är uppmuntrande med tanke på den framgång rapporterats från den senaste DREAM utmaning [7], där de bästa resultaten metod för synergi förutsägelse skulle ha tillåtit halvera storleken på screeningexperiment. Men det finns viktiga skillnader mellan vår studiedesign och av DREAM utmaning: DREAM analyserade transkriptom förändringar efter bredverkande kemoterapeutiska läkemedelsbehandlingar, medan vi undersökte effekten av inhibitorer med specifika mål, att förlita sig enbart på information från ostörda systemet <. br> i motsats till nätverksbaserade strategier, som ofta använder korrelationsanalys av storskaliga datamängder från olika fenotyper sjukdoms [45,46], eller storskaliga cellodlingsdrogstördata för att utbilda modeller för läkemedelssvar förutsägelser [6 , 7,9,47,48], utnyttjar vår modell-baserade strategi mekanistiska molekylära väg kunskap, tillgänglig i databaser, tillsammans med utgångsdata från de ostörda cancerceller i den valda experimentella system. Detta innebär att vår strategi möjliggör urval av intressanta kandidater för effektiv läkemedelskombinationer innan du utför själva läkemedelsstörnings experiment. Såvitt vi vet har detta inte varit framgångsrikt visat tidigare. De flesta av reglerande nätverk modellering fokus på signalering händelser som drivs av specifika hormonreceptorer. Detta gäller studier som undersöker logisk modellering för att förstå konsekvenserna av att störa specifika tillväxtfaktorn signalöverförings svar [16,18,49-51], liksom att kvantitativa och semi-kvantitativ modellering används för att förutsäga effekten av synergistisk signalöverföringsstörningar [6,31,52]. Däremot visar vår inställning att det är möjligt att effektivt använda en modell som representerar en cell öde beslut nätverk i aktivt växande celler utan att uttryckligen överväger någon extern tillväxtbefrämjande stimulans (t ex tillväxthormon). I själva verket, hävdar vi att använda attraktor av en fristående modell av en prolifererande cell som referenspunkt för läkemedelssynergi analys ger en god indikation på tillståndet hos aktivt växande cancerceller. Cancercellernas tillväxt anses drivas av en mångfald av tillväxtbefrämjande stimuli. Inte bara är den potentiella repertoaren av dessa signaler betydande, förhållandevis liten detalj om deras signaleringsmekanismer är känd. Vi antar därför att vi kan sammanfatta deras effekt genom att betrakta denna mångfald av signaler för att ge ett sammanhang främjar god tillväxt och att vi därför kan avfärda någon närmare. På grundval av detta rymmer vi en varaktig multifaktordriven proliferation [1] genom att använda en fristående modell, där alla komponenter som ingår regleras av andra noder i modellen. Konfigurationen av komponent aktiviteter kan sedan härledas från baslinjen biomarkörer som uppmätts i cancerceller. Tillsammans utgör dessa modell designprinciper gör det möjligt för oss att skapa en dynamisk modell anpassad till specifika cancerceller, men inte beroende av explicit extracellulärt input från specifika tillväxtbefrämjande medel ( e
biologiska kunskaper på intracellulära signalvägar med baslinjedata från AGS magsäcksceller. Konstruktionsprinciper vår analys styrs av antagandet att tillväxten av cancerceller till stor del driven av mekanismer som gör det möjligt dessa celler att utnyttja ett brett spektrum av tillväxtbefrämjande signaler från omgivningen. Denna aspekt av inre, ihållande multifaktordrivna cancer proliferation [1] tillgodoses genom att konstruera reglerande nätverk som en fristående modell: vi inkluderar endast noder som regleras av andra noder i modellen. Den valda designen undviker behovet av att modellera effekterna av specifika tillväxtfaktorreceptorer, med tanke på stället de integrerade svar från en mängd tillväxtfrämjande stimuli, som observerats vid bedömningen av aktiviteten av signalering enheter (proteiner och gener) som ingår i modellen. Av detta följer att de facto
tillväxtbefrämjande konfiguration av en sådan fristående modell kan fastställas genom att observera utgångs biomarkörer som uppmätts i cancercellerna.
Logisk modellering av adenocarcinom cell öde beslut
, ingår att representera cell öde fenotyper. Reglerings diagram med kommentarer finns i SBML format (se S1 datamängd och S1 tabell).
Konstruktion av en logisk modell.
, varje tar fyra värden (0, 1, 2, 3), och deras närmaste uppströms noder, caspase 3/7 och CCND1, var tar tre värden (0, 1, 2). Dessa multilevel variabla noder används endast för noder som styr utgångarna av modellen, och gjorde det möjligt att modellera graderade tillväxtbefrämjande /hämmande effekt (se Material och Metoder och S1 text). En logisk formel var associerat med varje komponent, som definierar hur dess aktivitetsnivå styrs av de av dess regulatorer. Vår standardmetod var att kombinera alla aktiverande regulatorer av ett mål med den booleska operatorn ELLER
och hämmande regulatorer av ett mål med operatören och inte rekommendera (som i [21]). Detta innebär att någon aktivator till fullo kan aktivera målnoden i frånvaro av inhiberande aktivitet. Vidare kan verkan av eventuell hämmande regulator fullständigt hämma målet, även i närvaro av aktiverande insignal från en eller flera aktivatorer. På grundval av biologisk kunskap och litteraturrapporter har mer specifika regler definieras för vissa delar av modellen (se S1 text). För β-catenin reaktionsvägen i synnerhet raffinerade vi logiska regler av noder som representerar aktiviteten av β-TrCP (den β-catenin förstörelse komplex), TCF (målet är β-catenin-aktivitet), och noden som representerar aktiviteten av β-catenin själv.
var vid sitt maximum, och Antisurvival vid sitt minimum, representerande stark proliferation ( Prosurvival
= 3, och Antisurvival
= 0). Denna modell stabilt tillstånd överensstämmer således med publicerade kunskap om molekylära tillstånd i aktivt växande AGS celler. Denna modell följs också med resultat från publicerade störnings experiment AGS-celler (se S1 Text och S6 tabell). Den resulterande logisk modell, kodad med programvaran GINsim v2.9, visas i figur 2. Motsvarande GINsim fil tillhandahålls som S2 dataset.
in silico
simuleringar förutsäga fem hämmare synergier
, genom att subtrahera värdet av Antisurvival
värdet av Prosurvival
avläsnings noder (varje flervärderad med tillstånd från 0 till 3), med ett värdeintervall från -3 till 3. Om attractor innehöll en unik stabilt tillstånd, beräkningen av tillväxt
var okomplicerad. När det gäller komplexa attraktorer använde vi medelvärdet av skillnaden Prosurvival Omdömen - Antisurvival
över alla stater som hör till attraktor. Vi sluta synergi när kombinationen av två hämmare producerade ett värde för tillväxt
lägre än det minsta värdet av hämmarna individuellt:
där perturbationN
är den störning av komponenten N
.
Experimentell utvärdering av modellprognoser
föreslår Modell en nyckelroll för FOXO i tillväxthämning synergi
Diskussion
. g
. Tillväxthormoner
 Hushållens desinfektionsmedel kan bidra till fetma hos barn
Hushållens desinfektionsmedel kan bidra till fetma hos barn
 Förlust av mikrobiom från antibiotikaanvändning påverkar svaret på influensavaccin
Förlust av mikrobiom från antibiotikaanvändning påverkar svaret på influensavaccin
 Mikrober kan förutsäga dödliga utfall hos ventilerade COVID-19-patienter
Mikrober kan förutsäga dödliga utfall hos ventilerade COVID-19-patienter
 Antibiotikaresistensen fördubblas på bara två decennier
Antibiotikaresistensen fördubblas på bara två decennier
 Studie med tvillingar visar att covid-19-symtom har ett genetiskt bidrag
Studie med tvillingar visar att covid-19-symtom har ett genetiskt bidrag
 Tarmmikrobiom förändras med tillagning av vegetabiliska livsmedel,
Tarmmikrobiom förändras med tillagning av vegetabiliska livsmedel,
 Bakteriofager kan behandla E. coli utan att skada tarmen,
säger ny studie Forskare i USA har utvecklat en ny terapi som använder en unik bakteriofag för att behandla E. coli -infektioner. Bakteriofagen befanns vara liknande i sin effektivitet mot infektion j
Bakteriofager kan behandla E. coli utan att skada tarmen,
säger ny studie Forskare i USA har utvecklat en ny terapi som använder en unik bakteriofag för att behandla E. coli -infektioner. Bakteriofagen befanns vara liknande i sin effektivitet mot infektion j
 Forskare utvinner fullständigt mänskligt genom från ett tusentals år gammalt "tuggummi"
Det verkar som att tuggummi inte är en ny trend! Forskare har hittat tuggummi som är 5, 700 år gammal och den har gett ledtrådar angående forntida DNA. En studie med resultaten publicerades i tidskrif
Forskare utvinner fullständigt mänskligt genom från ett tusentals år gammalt "tuggummi"
Det verkar som att tuggummi inte är en ny trend! Forskare har hittat tuggummi som är 5, 700 år gammal och den har gett ledtrådar angående forntida DNA. En studie med resultaten publicerades i tidskrif
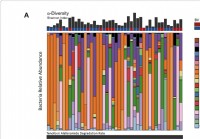 Det vaginala mikrobiomet kan påverka effekten av HIV -förebyggande behandling
En ny studie utförd av Dr Nichole Klatt från University of Minnesota, och kollegor, har avslöjat att vaginala mikrobiella samhällen är kopplade till en ökad risk för HIV -förvärv och kan också påver
Det vaginala mikrobiomet kan påverka effekten av HIV -förebyggande behandling
En ny studie utförd av Dr Nichole Klatt från University of Minnesota, och kollegor, har avslöjat att vaginala mikrobiella samhällen är kopplade till en ökad risk för HIV -förvärv och kan också påver