Estratto
caveolina-1 (CAV1) è una proteina impalcatura e del recettore patogeni nella mucosa del tratto gastrointestinale. L'infezione cronica delle cellule epiteliali gastriche da Helicobacter pylori Autore Sommario L'infezione con il batterio Helicobacter pylori Visto:. Hitkova Io, Yuan G, Anderl F, Gerhard M, Kirchner T, Reu S, et al. (2013) caveolina-1 Protegge B6129 i topi contro Helicobacter pylori Editor: Steven R. Blanke, University of Illinois, Stati Uniti d'America Ricevuto: 23 maggio 2012; Accettato: 4 febbraio 2013; Pubblicato: 11 apr 2013 Copyright: © 2013 Hitkova et al. Questo è un articolo ad accesso libero distribuito sotto i termini della Creative Commons Attribution License, che permette l'uso senza restrizioni, la distribuzione e la riproduzione con qualsiasi mezzo, a condizione che l'autore originale e la fonte sono accreditati Finanziamento:. Questo studio è stata sostenuta da sovvenzioni a favore di EB e MPAE dalla Deutsche Krebshilfe (108.287) e DFG (BU-2285). MPAE è supportato anche da finanziamenti della Deutsche Krebshilfe (107885), DFG (SFB 824, TP B1), Else Kröner Stiftung (Nr P14 /07 //A104 /06) e BMBF (MOBIMED 01EZ0802;. KMU-innovativ No 0315116B Il finanziatori avevano alcun ruolo nel disegno dello studio, la raccolta e l'analisi dei dati, la decisione di pubblicare, o preparazione del manoscritto Competere interessi:.. Gli autori hanno dichiarato che nessun interesse facente concorrenza esiste Introduzione Helicobacter pylori I due principali H. pylori caveolina-1 (CAV1) è il 21-24 kDa importante ed essenziale strutturale proteine di caveolae, una forma specializzata di microdomini zattera lipidica. Caveolae sono 50-100 pallone nm /invaginations a forma di tubo della membrana plasmatica abbondante nei macrofagi, cellule endoteliali e cellule muscolari lisce, di tipo I pneumociti e adipociti, dove partecipano i processi di trasporto cellulari tra cui l'endocitosi, efflusso di colesterolo e traffico di membrana [15] , [16]. In questo contesto, CAV1 può anche agire come un inibitore di endocitosi clatrina-indipendenti e bloccare patogeno /tossina assorbimento [17], [18]. Attraverso il legame al suo dominio ponteggi, CAV1 inibisce direttamente una pletora di recettori e gli enzimi tra cui tirosin-chinasi della famiglia Src e Ras, G-proteine e sintasi ossido di azoto [15]. Oltre ad un ruolo nel traffico di membrana, CAV1 costituisce quindi una piattaforma di controllo per la regolazione della proliferazione e sopravvivenza cellulare [19]. CAV1 esercita anche una funzione importante nella motilità cellulare e la migrazione e, all'interno epiteliali, stromali e tessuti endoteliali, facendo rispettare contatti cellula-cellula, l'adesione cellula-matrice e le risposte immunitarie [20], [21], [22], [23] . CAV1 lega direttamente il colesterolo, e trascrizione di CAV1 è regolata negativamente dal fattore di trascrizione steroli-reattiva elemento-binding protein-1 (SREBP1) [24]. SREBP1 è legato al reticolo endoplasmatico (ER) come inattiva 125 kDa precursore e si attiva in condizioni di carenza di colesterolo clivaggio proteolitico nel Golgi. Questa scissione è seguita dalla traslocazione del attiva 68 kDa SREBP1 nel nucleo dove si lega ad elementi steroli-reattiva (SRE) di geni target, tra cui CAV1, coinvolti nella sintesi di colesterolo e acidi grassi [25]. H. pylori Abbiamo quindi ipotizzato che il colesterolo-binding proteine SREBP1 e CAV1 sono bersagli di H. pylori Etica dichiarazione gli studi sugli animali sono stati condotti in accordo con le linee guida etiche della Technische Universität München (Germania Animal Welfare Act, Deutsches Tierschutzgesetz) ed era stato approvato (7.2-1-54-2531-74-08) per il governo della Baviera Animali omozigote CAV1 eliminazione diretta (CAV1-ko) (Cav1tm1Mls ceppo /J; magazzino numero 004.585) (Regierung von Obb, Monaco di Baviera, in Germania.). e abbinati selvatici controllo tipo (WT) (ceppo B6129SF2 /J; magazzino numero 101045) topi (8 settimane) sono stati ottenuti dal Jackson Laboratory (Bar Harbor, Maine) e mantenuto su uno sfondo misto in una struttura del mouse senza patogeni [28], [ ,,,0],29]. Sperimentale ulcera gastrica è stata eseguita con indometacina come pubblicato prima [30]. L'infezione dei topi con il CagA /VacA-consegna del mouse-adattato carente H. pylori prodotti chimici sono stati da Merck (Darmstadt, Germania) o Sigma (Taufkirchen, Germania). antisieri policlonali erano SREBP1 (# PA1-46142, Thermofisher scientifico, Waltham, MA), CAV1 (N-20, SC-894), SREBP1 (C-20, SC-366), CagA (B-300, SC-25766) , FAK (A-17, SC-557), fosfo-FAK (Tyr-397, SC-11765), Hsp90 alpha /beta (H-114, SC-7947), Lamin A /C (H-110, SC- 20681, il tutto da Santa Cruz Biotech., CA), generale e fosfo ERK1 /2 (p44 /p42), p38, JNK (il tutto da Cell Signaling, Danvers, MA) e Ki-67 (SP6, DCS GmbH, Amburgo, Germania ). Mouse anticorpi monoclonali sono stati CAV1 (ɢ.406) e fosfo-CAV1 (PY14,ɣ.338) (sia da BD /trasduzione Lab., San Jose, CA), DLC-1 (C-12, SC-271.915) e beta-actina (AC-15, SC-69879) (entrambi da Santa Cruz Biotech.). La specifica-macrofagi anti ratto del mouse-F4 /80 anticorpo (# MF48000) è stato ottenuto da Invitrogen (Life Technologies, Darmstadt, Germania). Pollo anti H. pylori rene embrionale umano (HEK293), Madin-Darby rene canino ( MDCK), linee di cellule umane GC parentali (AGS, MKN45, N87) (tutti dalla American Type Culture Collection, Rockville, MD) e cloni stabilmente trasfettati generati ne sono stati mantenuti, come descritto in precedenza [37]. L'infezione delle cellule con il CagA-consegna delle cellule-adattato abile H. pylori DNA-costrutti Il plasmide espressione pEGFP-CagA è stato menzionato altrove [38]. Il frammento ~800 bp del promotore prossimale CAV1 umana (AF019742, posizione 69-859) [24] è stato amplificato mediante PCR dal DNA genomico di fegato umano normale e clonato nei siti HindIII /KpnI di pGL3-Luc reporter luciferasi plasmide ( Promega GmbH, Mannheim, Germania). Isoforma 4 della umana DLC1 mRNA [39] (DLC1v4, NM_001164271.1) è stato amplificato da cellule di epatoma HepG2 umane e inseriti nei siti BamHI /NotI del vettore di espressione pTarget (Pt, Promega GmbH). saggi di trasfezione e luciferasi transitori sono stati eseguiti come in precedenza [37]. cultura batterica H. pylori ex vivo stomaci interi sono stati asportati dai topi, e la formazione di colonie è stato determinato essenzialmente come descritto [31] . Una striscia antrale dello stomaco è stato pesato, collocato in 5 ml di brodo e Brucella in agitazione per 10 min. Diluizioni di 1:10 1:100 e 1:1000 sono stati preparati, e 100 ml di ogni diluizione è stato placcati su H. pylori Lo stomaco residuo è stato lavato con acqua sterile. Una striscia antrale è stato tagliato, congelati in azoto liquido e conservati a -80 ° C fino estrazione di RNA. Il resto dello stomaco è stata posta in 3 ml di 4% (w /v) paraformaldeide (PFA) in tampone fosfato isotonico (PBS) e incubate per 24 ore a 4 ° C. Poi, lo stomaco è stato tagliato lungo la curvatura più grande e piccolo in due metà, seguita da disidratazione e inclusione in paraffina per l'analisi istologica. Le cellule sono state infettate con il H. pylori rilevamento di proteine immunoprecipitate di SDS-PAGE e WB è stata eseguita come prima [41]. Laser desorbimento /ionizzazione spettrometria di massa Matrix-assistita (MALDI-MS) è stato descritto in dettaglio in [29]. La colorazione è stata eseguita in modalità triple-colore visualizzazione 4,6- diamidino-2-phenylindole (DAPI), Alexa-488 e -594 utilizzando una fotocamera digitale collegata (AxioVision, rilasciano 4.4) microscopio a fluorescenza (Axiovert 200M, Carl Zeiss MicroImaging GmbH, Hallbergmoos, Germania). microscopia confocale (Axiovert 40, Zeiss) e 3D-ricostruzione di H. pylori La valutazione istopatologica e immunoistochimica (IHC) cronica attiva gastrite è stata definita dalla presenza contemporanea di entrambi polymorphnuclear neutrofila (PMN) e cellule mononucleate (linfociti e plasmacellule) all'interno della mucosa gastrica. Attivo (PMN) e cronica (mononucleate) infiltrato è stata valutata come segue: tessuto gastrico paraffina-embedded è stato tagliato in 3 sezioni micron utilizzando un microtomo semiautomatico (Leica Microsystems GmbH, Wetzlar, Germania). Le sezioni sono state poi colorate con ematossilina & Eosina (H & E) soluzioni. L'analisi istopatologica è stata effettuata da tre patologi (CR, SR, TK) in cieco per l'impostazione di studio. alterazioni morfologiche della mucosa gastrica sono stati classificati secondo il sistema Sydney aggiornato [32], [42]. Il grado di gastrite è stato segnato basata sulla densità della infiltrati infiammatori intramucoso da mononucleate e cellule PMN come pubblicato prima [43]: nessuno (0), lieve (1+), moderata (2+) e grave (3+). Inoltre, iperplastico o alterazioni epiteliali rigenerative, perdita di cellule parietali e la frequenza dei follicoli linfoidi o aggregati linfoidi sono stati notati. L'intensità di H. pylori Chip (Kit da Upstate, Millipore GmbH, Schwalbach, Germania) e tutti gli altri metodi sono stati eseguiti come descritto in precedenza [45]. Oligonucleotidi sono elencati nella Tabella S1. La vitalità delle cellule aderenti è stata misurata mediante 1- (4,5-dimetiltiazol-2-il) 3,5-difenil-formazano (MTT ) test (Roche Diagnostics GmbH, Mannheim, Germania) come raccomandato dal produttore. Per determinare l'adesione cellulare, 1 × 10 4 cellule sono state seminate in 6 cm piatti di coltura cellulare da 1 a 6 h seguita da lavaggio ripetuto con PBS. Le cellule aderenti rimanenti sono state fissate con 4% (w /v) in PBS PFA, colorate con cristalvioletto e successivamente contati utilizzando ImageJ (NIH, Bethesda, MD). saggi di guarigione della ferita sono stati effettuati essenzialmente come descritto in [46]. Brevemente, le cellule sono state coltivate a confluenza in 6 piatti cm, e un graffio 5 millimetri è stato introdotto nel monostrato utilizzando una punta blu rovesciata seguita da incubazione delle piastre di coltura cellulare per ulteriori 24, 48 e 72 h. la chiusura della ferita è stato monitorato su fissazione e colorazione delle cellule con cristallo viola utilizzando la microscopia in campo chiaro (Axiovert 200M, Carl Zeiss MicroImaging GmbH). Statistiche I risultati sono mezzi ± S.E. da almeno 5 animali per genotipo o 3 esperimenti indipendenti provenienti da diversi passaggi in coltura. Il software GraphPad Prism (versione 4.0, La Jolla, CA) è stato utilizzato per analizzare i dati. P-valori (* p < 0,05) sono stati calcolati utilizzando t e test esatto di Fisher di Student numeri di adesione Umano: CAV1:. NM_001753.4, Q03135; B2M: NM_004048.2, P61769; IL8: NM_000584.3, P10145; DLC1 v1: NM_182643.2, Q96QB1; DLC1 v4: NM_001164271.1, Q96QB1; ACS: NM_018677.3, Q9NR19; HMGCoAS: NM_001098272.2, Q01581; HMGCoAR: NM_000859.2, P04035; LDLR: NM_000527.4, P01130; beta-Actina: P60709; Lamina A: P02545; Lamin C: P02545; alpha Hsp90: P07900; beta Hsp90: P08238; ERK1 (P44): P27361; ERK2 (p42): P28482; FAK: Q05397; JNK1: P45983; JNK2: P45984; p38: Q16539; Src: P12931; SREBP1: P36956; Ki-67: P46013; Mouse: CAV1: NM_007616.4, P49817; B2M: NM_009735.3, Q91XJ8; TNFalpha: NM_013693.2, P06804; IFNgamma: NM_008337.3, P01580; IL1beta: NM_008361.3, P10749; IL6: NM_031168.1, P08505; CD4: NM_013488.2, P06332; CD19: NM_009844.2, P25918; CD25: NM_000417.2, P01589; CD86: NM_019388.3, P42082; CCL5: NM_013653.3, P30882; CXCL1: NM_008176.3, P12850; PPARG2: NM_015869.4, P37231; TFF2: NM_009363.3, Q9QX97; Cane: B2M: NC_006612, XP_850148; H. pylori Risultati topi CAV1-deficienti display migliorato gastrite dopo l'infezione con CagA-consegna incompetente H. pylori Per valutare i cambiamenti istologici indotti nel tessuto gastrico su H. pylori CAV1 carenza favorisce il reclutamento di macrofagi nella mucosa gastrica infetto Per valutare l'identità delle cellule immunitarie che contribuiscono a H. pylori Risultati simili sono stati ottenuti da esperimenti di introduzione di una rapida lesioni gastriche nei topi mediante iniezione di indometacina [30] (Fig.S1). Coerentemente con il danno tissutale rafforzata in stomaco CAV1-KO (* p = 0,0161, WT contro Per valutare la funzione di CAV1 durante H. pylori CAV1 protegge le cellule umane GC contro riarrangiamento CagA-indotta del citoscheletro La formazione di proiezioni aghiformi ( "ronzio uccello") è un tipico fenotipo morfologica delle cellule AGS in risposta alle infezioni con CagA-consegna abile H. pylori A sostegno di questi risultati, l'adesione cellulare e tassi di chiusura della ferita sono stati più pronunciati in AGS /CAV1 rispetto alle cellule AGS /EV (Fig.S2). Coerentemente con la sua funzione di una proteina bersaglio di CagA e componenti di adesioni focali [48], analisi WB (Fig. 4D) anche rilevato livelli più alti (0,4 ± 0,1 AGS /EV contro CagA-consegna competente H. pylori CAV1 ha dimostrato di essere fosforilata da citosolici tirosin-chinasi (Src, Abl) in tirosina 14 [49], e fosforilata CAV1 e src sia attivare le piccole GTPasi Rho /Rac /Cdc42 che regolano le funzioni del citoscheletro [13], [50]. Per identificare il meccanismo molecolare alla base come CAV1 protegge contro CagA-stress legato cellule, abbiamo valutato la segnalazione di percorsi avviati da CagA-consegna abile H. pylori Non è stato possibile rilevare una interazione diretta o colocalization quantitativa di CagA proteine o H. pylori Per identificare una proteina candidato che conferisce protezione CagA in un CAV1-dipendente modo, è stata eseguita una schermata di interazione proteina basato su MALDI-MS (Fig. 7A). cellule AGS /CAV1 sono state infettate per 16 h con H. pylori Questo risultato ci ha spinto ad amplificare il cDNA di variante 4 di DLC1 umana [39] da cellule HepG2 epatoma umano (fig . 7C). Il cDNA è stato inserito nel vettore di espressione pTarget (pT-DLC1v4) seguita da trasfezione transiente in AGS parentali o cellule HEK293 per 24 h. WB analisi ha rilevato l'espressione di una proteina ~110 kDa, coerente con le dimensioni previsto di DLC1v4 [39]. cellule AGS transitoriamente trasfettate sono stati poi infettati con H. pylori
( H. pylori
) è un importante fattore di rischio per il cancro gastrico umano (GC) dove CAV1 è spesso down-regolato. Tuttavia, la funzione di CAV1 in H. pylori
infezione e patogenesi della GC è rimasta sconosciuta. Mostriamo qui che i topi CAV1-deficienti, infettate per 11 mesi con la CagA-consegna carenti H. pylori
ceppo SS1, ha sviluppato più gravi gastriti e danni ai tessuti, inclusa la perdita di cellule parietali e iperplasia foveolare, e visualizzato in basso la colonizzazione della mucosa gastrica di wild-type cucciolata B6129. topi CAV1-nulli mostrato migliorata infiltrazione di macrofagi e cellule B e la secrezione di chemochine (RANTES) ma avevano ridotto i livelli di CD25 + cellule T regolatorie. cellule CAV1-deficienti umani GC (AGS), infettati con il CagA-consegna abile H. pylori
ceppo G27, erano più sensibili alle morfologie di stress del citoscheletro CagA-correlate ( "ronzio uccello") rispetto alle cellule AGS stabilmente trasfettate con CAV1 (AGS /CAV1). L'infezione di cellule AGS /CAV1 innescato l'assunzione di p120 RhoGTPase-attivazione di proteine /cancellati nel cancro al fegato-1 (p120RhoGAP /DLC1) per CAV1 e neutralizzato riarrangiamenti del citoscheletro CagA-indotta. Nelle linee cellulari umane GC (MKN45, N87) e il tessuto del mouse stomaco, H. pylori
espressione endogena down-regolato di CAV1 indipendentemente CagA. Meccanicamente, H. pylori
attivato steroli-reattiva elemento-binding protein-1 (SREBP1) per reprimere la trascrizione del gene umano CAV1 da elementi steroli-reattiva (SRE) nel promotore prossimale CAV1. Questi dati suggeriscono un ruolo protettivo di CAV1 contro H. pylori
indotta infiammazione e danni ai tessuti. Proponiamo che H. pylori
exploit down-regulation di CAV1 per sovvertire la risposta immunitaria dell'ospite e di promuovere la segnalazione dei suoi fattori di virulenza in cellule ospiti.
( H. pylori
) colpisce soprattutto i bambini nei paesi in sviluppo che sono a rischio di progredire a cancro gastrico (GC), come gli adulti, dopo molti anni di infezione persistente, in particolare con i ceppi che sono positivi per il fattore di virulenza oncogenico CagA. Eradicazione di H. pylori
da antibiotici è un trattamento di scelta, ma può anche alterare la suscettibilità alle allergie e di altri tipi di tumore. Così, nuovi marcatori diagnostici o prognostici sono necessari che rilevano cambiamenti molecolari precoci della mucosa dello stomaco durante il passaggio di infiammazione cronica al cancro. Nel nostro studio, abbiamo scoperto che il soppressore del tumore caveolina-1 (CAV1) è ridotto dopo l'infezione con H. pylori
, e CagA era sufficiente ma non necessaria per questo down-regulation. La perdita di CAV1 è stato causato da H. pylori
attivazione-dipendente di steroli-sensibile elemento-binding protein-1 (SREBP1), e questo evento abolito l'interazione di CAV1 con p120 RhoGTPase-attivando proteina /cancellato nel cancro al fegato-1 (p120RhoGAP /DLC1), un secondo in buona fede
soppressore del tumore nel tessuto gastrico. Conclusivamente, CAV1 e DLC1 possono costituire marcatori molecolari del H. pylori
mucosa gastrica -infected prima trasformazione neoplastica dell'epitelio
gastrite. PLoS Pathog 9 (4): e1003251. doi: 10.1371 /journal.ppat.1003251
( H. pylori
) è un batterio Gram-negativo che colonizza lo stomaco di ca. il 50% della popolazione mondiale e aumenta il rischio per lo sviluppo di cronica gastrite, ulcera peptica, gastrica associato alla mucosa tessuto linfoide (MALT) linfoma, atrofia della mucosa e cancro gastrico (GC) [1], [2]. sulla base di questa eziologia, H. pylori
è stato classificato come classe I agente cancerogeno dall'Organizzazione mondiale della sanità (OMS) nel 1994 [3].
tossine [4], CagA e VacA, vengono interiorizzati in gastrica cellule epiteliali per iniezione tramite il tipo batterico sistema di secrezione IV (CagA) [5] o con inserimento diretto in raft lipidici (VacA) [6], [7]. raft lipidici sono colesterolo e ricchi di sfingolipidi microdomini della membrana plasmatica [8], [9], che sono sfruttate da molti agenti patogeni, tra cui virus, parassiti e batteri, per facilitare la diffusione di organismi interi e /o internalizzazione di tossine nelle cellule ospiti [ ,,,0],10], [11], [12]. Ad esempio, Neisseria spec
. utilizza zattere lipidiche e segnalazione Rho-mediata del citoscheletro actina per accedere al citosol [13]. Pseudomonas aeruginosa
sfrutta lipidi cellule epiteliali zattera-associata recettore Toll-like 2 per l'infezione del polmone [14].
ha dimostrato di metabolizzare il colesterolo dalla membrana della cellula ospite, e colesterolo ospite altera le proprietà oncogeniche di CagA [26], [27].
funzioni di infezione e /o effettori. In particolare, abbiamo chiesto se (i) H. pylori
exploit CAV1 per facilitare l'iniezione e la segnalazione a valle di CagA nelle cellule epiteliali gastriche o (ii) CAV1 agisce come una proteina protettiva "barriera-enforcing" che contrasta la malattia evocato da H. pylori
. Per verificare ciò, i fenotipi derivanti da H. pylori
infezione sono stati studiati in topi CAV1-carenti e in linee cellulari umane GC. I nostri dati hanno mostrato che i topi CAV1 protetto B6129 contro H. pylori
-related gastriti e danni ai tessuti in vivo
indipendentemente CagA. H. pylori
attivato anche SREBP1 e l'espressione down-regolato di murine e CAV1 umana indipendentemente CagA. Inoltre, CAV1 contrastato riarrangiamenti del citoscheletro CagA-dipendenti in vitro
da assunzione del soppressore del tumore eliminato nel fegato cancro-1 (DLC1).
Materiali e Metodi
ceppo SS1 è stata eseguita mediante sonda gastrica, come descritto [31]. I topi tempo medio di diversa provenienza genetici (C57BL /6, B6129, BALB /c) prendere per progredire a gastrite cronica e oltre (atrofia gastrica, iperplasia, displasia) [32] varia tra 10 e 15 mesi dopo l'infezione con il riferimento standardizzato ceppo SS1 [28], [33], [34], [35]. Abbiamo quindi deciso di effettuare la nostra analisi in questo lasso di tempo.
Reagenti
policlonale Ab è stato utilizzato come descritto [36]. citochine siero sono stati misurati mediante test ELISA (R & D Systems, Minneapolis, MN) secondo le istruzioni del produttore. Pull-down test per le piccole GTPasi Rho /Rac /Cdc42 sono stati acquistati da Biocat (Heidelberg, Germania).
Cell cultura
G27 ceppo è stato eseguito come prima [36].
SS1 e batteri G27 sono stati recuperati dalle scorte C glicerolo -80 ° e cresciuto su Wilkins-Chalgren (WC) piastre di agar sangue in condizioni microaerofilia (10% di CO2, 5% di O2, 85% N2; 37 ° C) per 2-3 giorni. Il H del mouse-adattato. pylori
SS1 è stato raccolto da piastre di agar per in vivo
infezioni come pubblicato in precedenza [31]. Il ceppo era SS1-PCR positiva per il cagA
gene e mRNA ma non iniettare proteina CagA funzionale [40] come evidente dall'assenza del fenotipo "ronzio uccello" in cellule infettate AGS (dati non mostrati) . Il H cell-adattato. pylori CagA-
batteri consegna abile G27 peso
e la CagA-delezione mutante G27 Delta cagA
sono state raccolte da piastre di agar e successivamente coltivata in continuo co-coltura con cellule MDCK come descritto [36].
quantificazione di unità formanti colonie (CFU)
piastre di agar sangue WC -selettivo. Il numero di colonie batteriche è stata determinata dopo 5 giorni e normalizzati al peso dei pezzi stomaco corrispondenti.
Lavorazione di topo gastrica tessuto
Gentamicina saggio di protezione
ceppo G27 per 2 a 24 ore a una molteplicità di infezione (MOI) di 500:1. Successivamente, le cellule sono state lavate tre volte con PBS per rimuovere batteri residui e sono stati ulteriormente incubati per 2 ore a 37 ° C in atmosfera umidificata in DMEM /F12 (10% FCS, 10% Brucella brodo) integrato con gentamicina (200 mg /ml ), penicillina /streptomicina (100 ug /ml) e cloramfenicolo (100 mcg /ml). L'assenza di batteri extracellulari è stato confermato al microscopio, e le cellule sono state successivamente lisate per il rilevamento di CagA intracellulare da Western Blot (WB).
Coimmunoprecipitation (COIP) e Western Blot (WB)
immunofluorescenza
cellule -infected con LSM510 (Zeiss) e Volocity (Improvision, Tubinga, Germania) è stato fatto come prima [37].
colonizzazione della mucosa gastrica è stato registrato come lievi (batteri pochi e singoli in una distribuzione casuale), moderato (batteri singoli e raggruppati in una distribuzione discontinua) e grave (cluster batteriche densi che ricoprono la mucosa gastrica in strati continui). colonne multiple di diverse regioni dello stomaco sono stati determinati. Immunoistochimica (IHC) è stata eseguita su sezioni di paraffina come descritto in precedenza [44].
elettroforetica saggio mobility shift (EMSA), immunoprecipitazione della cromatina (ChIP), trascrizione inversa PCR (RT-PCR) e PCR quantitativa (qPCR)
saggi cellulari
: CagA: YP_002266135.1, B5Z6S0; UreB:. YP_626814.1, Q1CV82
SS1
infezione, B6129 WT e topi CAV1-KO sono stati infettati con il mouse-adattato e CagA-consegna carenti H. pylori
ceppo SS1. I topi sono stati sacrificati 11 mesi più tardi, e H. pylori
è stato isolato dal tessuto dello stomaco asportato [31]. topi CAV1-KO ha mostrato la colonizzazione batterica meno della mucosa gastrica rispetto ai topi WT (7.3 ± 2.4 WT contro
1,6 ± 0,5 KO × 10 3 CFU /mg tessuto dello stomaco; * p = 0,0141; n = 15 per genotipo) (Fig. 1A). analisi istopatologica ha rivelato che i topi WT e sia CAV1-KO sviluppati gastrite cronica attiva accompagnata da infiltrazione di cellule polymorphnuclear (PMN) mononucleari e nella mucosa gastrica (Fig. 1B). Al contrario, i topi WT e CAV1-KO non infettati avevano infiammazione intramucosa (dati non mostrati). Invece, la gastrite è stata notevolmente migliorata in H. pylori
-infected topi CAV1-KO rispetto ai topi WT infetti (Fig. 1C). Nei topi CAV1-KO, il punteggio medio di gastrite (0,7 ± 0,2 WT contro
1,7 ± 0,1 KO; * p = 0.0002, n = 15 per genotipo) è stato più grave (Tabella 1) rispetto ai topi WT , e la mucosa dello stomaco esposto intramucoso follicoli a cellule B, iperplasia foveolare e la perdita di cellule parietali. Questi dati indicano che CAV1-carenza è associata con un aumento della risposta infiammatoria nella mucosa gastrica e una colonizzazione meno efficiente da H. pylori
.
infiammazione -related nei topi CAV1-KO, analisi RT-qPCR di citochine selezionati, marcatori di superficie e chemochine è stata effettuata (Fig. 2A). Coerentemente con l'infiammazione osservata, H. pylori
espressione SS1 indotta di TNFalfa e IFNgamma nella mucosa gastrica di entrambi i topi WT e KO. Inoltre, abbiamo dichiarato un aumento dell'espressione di mRNA di CD19 (cellule B) (1.6 ± 0.3 WT contro
3,3 ± 0,9 KO; p = 0,0512; n = 15 per genotipo) e RANTES (CCL5) (1.3 ± 0.2 WT contro
2,1 ± 0,6 KO; p = 0,0449; n = 15 per genotipo) nel tessuto gastrico di H. pylori
-infected topi CAV1-KO rispetto ai topi WT infetti. Al contrario, i livelli di mRNA di CD4 (cellule T-helper), CD25 (cellule T-normativi) e CD86 (cellule presentanti l'antigene) sono stati soppressi da H. pylori
indipendentemente dallo stato CAV1. Immunoistochimica (IHC) ha rilevato un marcato aumento di intramucoso F4 /macrofagi 80-positivi nel tessuto gastrico di topi CAV1-KO infette rispetto al WT cucciolata (Fig. 2b). linfociti CD3 positivi erano situati intorno e dentro di follicoli intramucoso (dati non riportati).
KO, n = 9 per genotipo), caratterizzata da infiammazione, erosione e ulcere, i topi deficienti CAV1-anche espresso importi superiori di mRNA codificanti per la guarigione dell'ulcera proteine trifoglio fattore-2 (TFF2) (0.8 ± 0.3 WT contro
2.3 ± 0.4 KO; * p = 0,0048; n = 9 per genotipo) e dei perossisomi recettore attivante la proliferazione gamma (PPARg) (0.6 ± 0.2 WT contro
2,5 ± 0,5 KO; * p = 0.0008; n = 9 per genotipo). In sintesi, questi dati indicano che la perdita di CAV1 aumenta la sensibilità dei topi per gastrica infiammazione e danni ai tessuti.
CAV1 né adesione altera di H. pylori
ceppi né la sopravvivenza delle cellule umane GC
infezione in vitro
, la linea umana gastriche epiteliali cellule AGS è stata utilizzata che era stato stabilmente trasfettate con CAV1 plasmide di espressione (AGS /CAV1) o vuoto vettore (AGS /EV) [37]. In primo luogo, abbiamo esaminato se CAV1 influenza la sopravvivenza cellulare su H. pylori
infezione (Fig. 3A). cloni AGS con e senza CAV1 sono state infettate per 48 ore con il competente H CagA-consegna delle cellule-adattato. pylori
ceppo G27 a diverse molteplicità di infezione (MOI) che vanno da 1:100 a 1:2000. saggi MTT colorimetrici rivelato che CAV1 ha avuto alcun effetto sulla sopravvivenza globale delle cellule AGS su H. pylori
infezione. Risultati simili sono stati ottenuti con CagA-consegna incompetente H. pylori
SS1 e da Western Blot (WB) rilevare l'espressione e la fosforilazione delle chinasi di sopravvivenza (AKT /PKB, ERK1 /2, p38MAPK) (dati non riportati). Dal momento che sia H. pylori
e CAV1 interagire all'interno raft lipidici, abbiamo chiesto se l'adesione dei batteri alle cellule dipende dalla presenza di CAV1. AGS /CAV1 e cellule /EV AGS stati infettati (MOI = 10) con G27 (Fig. 3B, C) o SS1 (dati non mostrati) batteri per 30 minuti, seguita da lavaggio e successiva incubazione in mezzo fresco per 2 ore. Successivamente, le cellule sono state colorate per immunofluorescenza, e il numero di batteri che hanno aderito al CAV1 esprimono o vuoti cellule transfettate vettori sono stati contati (Fig. 3B, C). Non sono state osservate differenze di adesione tra AGS /CAV1 e le cellule AGS /EV, il che suggerisce che CAV1 non influenza l'adesione di H. pylori
batteri alle cellule ospite.
ceppi e traslocazione di CagA nel citoplasma [47]. Per esaminare il ruolo di CAV1 in questo riarrangiamento indotta da stress del citoscheletro, AGS /CAV1 e AGS /EV sono stati infettati per 16 h con H. pylori
G27 peso
o il mutante isogenico Delta cagA
(MOI = 100). Le cellule infettate sono state colorate come descritto sopra, e il numero di cellule AGS allungate sono stati determinati (Fig. 4A, B). cellule CAV1-deficienti AGS mostrato morfologie molto più allungate rispetto alle cellule che esprimono CAV1-(11 ± 0,8% AGS /EV contro
4 ± 0,8% AGS /CAV1; * p = 1,1 × 10 -8; n = 3, per clone). Come previsto, non fenotipo "colibrì" è stato ottenuto in cellule infettate con il CagA-consegna carenti SS1 o CagA-delezione mutante G27 Delta CagA
ceppi che sono entrambi in grado di iniettare la proteina CagA funzionale nelle cellule ospiti (dati non mostrati). cellule AGS /EV prodotte anche più IL8 mRNA su H. pylori
infezione G27 di cellule AGS /CAV1 (64 ± 19 EV contro
19 ± 6 CAV1; * p = 0,0176; n = 3, per clone) (Fig. 4C). Questi dati hanno indicato che CAV1 protegge contro CagA-stress legato cellule.
1,4 ± 0,1 AGS /CAV1 , * p = 0,0012; n = 3, per clone) di fosforilata adesione focale chinasi (FAK) in CAV1 che esprimono le cellule infettate con H. pylori
G27. Questi dati corroborato che le cellule AGS /CAV1 infettati con CagA-consegna competente H. pylori
mantenuto la loro forma epiteliale spread-out rispetto al fenotipo allungata sottolineato di cellule CAV1 /EV.
G27 innesca legame di p120RhoGAP /DLC1 a CAV1 nelle cellule umane GC
G27. L'infezione delle cellule AGS evocato una rapida fosforilazione di CAV1 nelle cellule AGS /CAV1 e di Src in entrambe le cellule /CAV1 e AGS /EV AGS. Questo risultato indicava che CAV1 agisce valle dell'attivazione Src CagA-dipendente, ma a monte della attivazione delle piccole GTPasi (Fig. 5A, B). Coerentemente con questa conclusione, i livelli di proteina di JNK fosforilata, che risiede al di sotto di Src, sono stati superiori a /EV cellule AGS rispetto alle cellule AGS /CAV1.
batteri G27 con CAV1 in COIP o esperimenti di immunofluorescenza (Fig. 6A, B). saggi di protezione gentamicina hanno rivelato che la quantità totale di CagA intracellulare iniettato era anche indipendente dalla presenza di CAV1 (Fig. 6C). Così, CAV1 né adesione inibito di H. pylori
batteri agli né iniezione di CagA nella cellula ospite, ma piuttosto ridotti gli effetti a valle del CagA su segnalazione intracellulare.
G27 (MOI = 100) seguita da lisi delle cellule a temperatura ambiente in MES tamponata 1% (v /v) Triton-X100. bande proteina precipitata da CAV1 antisiero sono stati visualizzati mediante colorazione argento e peptidi sono stati identificati mediante MALDI-MS, come pubblicato in precedenza [29]. Un frammento della proteina di ~95 kDa conteneva peptidi corrispondente alla variante 4 di p120 Rho proteina gap /cancellato nel fegato cancro-1 (p120RhoGAP /DLC1) [51], [52], un soppressore del tumore associato con adesioni focali e caveolae /raft lipidici [53]. DLC1 variante 4 (DLC1v4) ha una dimensione prevista di ~110 kDa ed è stato arricchito nei campioni di cellule che erano stati infettati con H. pylori
G27 rispetto alle cellule non infette (Tabella S2). Questi risultati sono stati confermati da COIP di CAV1 e endogena di proteine nelle cellule DLC1 AGS /CAV1 (Fig. 7B), indicando che H. pylori
G27 evocato un reclutamento specifico di DLC1 per CAV1 nelle cellule epiteliali gastriche umane infettate.
G27 (MOI = 100) per ulteriori 16 ore. Immunofluorescenza ha rivelato che DLC1 di per sé
non inibire la formazione del fenotipo CagA-indotta "colibrì" (19 ± 2% AGS /DLC1 contro
19 ± 2% AGS /EV; n = 3 per clone) rispetto a celle vuote vettore transfettate (Fig. 8A, B). Invece, DLC1 promosso cellule diffusione (20 ± 3% AGS /DLC1 contro
11 ± 2% AGS /EV; * p = 0,0067; n = 3, per clone) coerente con il suo ruolo nella regolazione delle adesioni focali [ ,,,0], H. H. pylori
infezione. pylori
.
 Come si chiama un medico che cura problemi digestivi?
Come si chiama un medico che cura problemi digestivi?
 Il microbioma del pene è un serbatoio per i batteri associati alla vaginosi batterica
Il microbioma del pene è un serbatoio per i batteri associati alla vaginosi batterica
 citrato di magnesio
citrato di magnesio
 Che cos'è la medicina funzionale?
Che cos'è la medicina funzionale?
 Suggerimento per la salute:tenere sotto controllo la sindrome dell'intestino irritabile
Suggerimento per la salute:tenere sotto controllo la sindrome dell'intestino irritabile
 5 frullati per l'aumento di peso del cibo reale (Paleo e SCD)
5 frullati per l'aumento di peso del cibo reale (Paleo e SCD)
 La bugia senza glutine:perché la maggior parte dei celiaci muore lentamente
La medicina convenzionale di solito funziona così... Ho un problema, il dottore capisce qual è il problema e mi dà una ricetta convenzionale generalmente supportata da medici, ricercatori e FDA. Que
La bugia senza glutine:perché la maggior parte dei celiaci muore lentamente
La medicina convenzionale di solito funziona così... Ho un problema, il dottore capisce qual è il problema e mi dà una ricetta convenzionale generalmente supportata da medici, ricercatori e FDA. Que
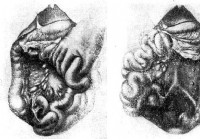 Diagnosi di linfoadenite mezenterialny acuta - Diagnosi di addome acuto
DIAGNOSI DI LABORATORIO Leucocitosi a mezenterialny lymphadenitis di solito alto — ai nostri pazienti che vanno da 9000 a 28.000 che anche caratterizza questa malattia come processo di carattere tutto
Diagnosi di linfoadenite mezenterialny acuta - Diagnosi di addome acuto
DIAGNOSI DI LABORATORIO Leucocitosi a mezenterialny lymphadenitis di solito alto — ai nostri pazienti che vanno da 9000 a 28.000 che anche caratterizza questa malattia come processo di carattere tutto
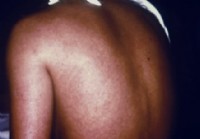 Malattia da virus di Marburg
Fatti che dovresti sapere sulla malattia da virus di Marburg Immagine:una donna con infezione da virus Marburg ha uneruzione cutanea sulla schiena; FONTE:CDC La malattia del virus di Marburg è endemi
Malattia da virus di Marburg
Fatti che dovresti sapere sulla malattia da virus di Marburg Immagine:una donna con infezione da virus Marburg ha uneruzione cutanea sulla schiena; FONTE:CDC La malattia del virus di Marburg è endemi