Фон H. Pylori Выводы Введение хеликобактерной Материалы и методы Пациенты <бр> H. штаммы пилори нуклеотидную последовательность Данные нуклеотидные последовательности, описанные здесь, доступны под DDBJ номерами доступа AB923031 к AB923492. Результаты В этом исследовании мы включили H. штаммы пилори Вывод Выражение признательности
<р> Недавний отчет показал, что филогенетическое происхождение хеликобактерной
на основе полилокусной последовательности ввода (MLST) была в значительной степени связано с выраженность гастрита в Колумбии. Тем не менее, потенциал отношений между филогенетического происхождения и клинических исходов не было рассмотрено в этом исследовании. Если филогенетическое происхождение, а не факторы вирулентности были действительно связаны с клиническими результатами, идентификации населения с высоким риском развития рака желудка в Колумбии было бы относительно просто. В данном исследовании мы исследовали филогенетические происхождение штаммов от рака желудка и язвенной болезни двенадцатиперстной кишки, живущих в Боготе, Колумбия.
Методы
<р> Мы включили 35 больных раком желудка и 31 больных язвенной болезнью двенадцатиперстной кишки, которые рассмотрены результаты варианта. Генотипы CaGa
и Вака
определяли с помощью полимеразной цепной реакции. Генеалогия этих колумбийских штаммов анализировали с помощью MLST. Бактериальная структура населения была проанализирована с помощью программного обеспечения структуры.
Результаты
штаммы от рака желудка и язвенной болезни двенадцатиперстной кишки были разбросаны в филогенетического дерева; Таким образом, мы не обнаружили никакой разницы в филогенетического распределения между раком желудка и двенадцатиперстной кишки в штаммах группе hpEurope в Колумбии. Шестьдесят шесть штаммов, с одним исключением, были классифицированы как hpEurope независимо от CaGa
и Вака
генотипов и типа заболевания. Структурный анализ показал, что колумбийские штаммы hpEurope имеют филогенетическое соединение испанских штаммов.
<р> Наше исследование показало, что филогеографического происхождение определяется MLST было недостаточно для проведения различий между раком желудка и двенадцатиперстной кишки риск язвенной болезни среди hpEurope штаммы в регионе Анд в Колумбии. Наш анализ также показывает, что hpEurope штаммы в Колумбии были введены в первую очередь испанских иммигрантов
<р> Цитирование:. Shiota S, Suzuki R, Мацуо Y, Miftahussurur M, Tran ТТХ, Бинь ТТ и др. (2014) хеликобактер пилори
от рака желудка и двенадцатиперстной кишки Показать То же филогеографического происхождения в Андском регионе в Колумбии. PLoS ONE 9 (8): e105392. DOI: 10.1371 /journal.pone.0105392
<р> Редактор: Масару Като, Национальный центр рака, Япония
<р> Поступило: 7 марта 2014; Принято: 23 июля, 2014 года; Опубликовано: 14 августа 2014
<р> Copyright: © 2014 Shiota и др. Это статья с открытым доступом распространяется в соответствии с условиями лицензии Creative Commons Attribution, которая позволяет неограниченное использование, распространение и воспроизведение на любом носителе, при условии, что оригинальный автор и источник приписывают наличие
<р> Data:. авторы подтверждают, что все данные, лежащие в основе выводы полностью доступны без ограничений. Данные нуклеотидную последовательность, описанные здесь, доступны под DDBJ номерами доступа AB923031 к AB923492
<р> Финансирование:. Этот доклад основан на работе, поддержанной частично за счет грантов от Национального института здоровья (DK62813) (YY) и гранты -в-помощи для научных исследований Министерства образования, культуры, спорта, науки и технологии (МПКСНТ) Японии (22390085, 22659087, 24406015 и 24659200) (YY), (23790798) (СС) и специальные координационные фонды для содействия Наука и техника из Японского агентства по науке и технике (JST). Доноры не играет никакой роли в дизайн исследования, сбора и анализа данных, решение о публикации или подготовки рукописи
<р> Конкурирующие интересы:.. Авторы заявили, что не существует никаких конкурирующих интересов
представляет собой спираль, грамотрицательная бактерия, которая инфицирует более половины населения мира [1]. Механизм передачи H. пилори
не выяснена, но распространение вируса от человека к человеку через орально-оральным или фекально-оральным путем считается наиболее вероятным [2]. H. Pylori
инфекция теперь принято быть связаны с серьезными гастрит-ассоциированных заболеваний, в том числе язвенной болезни и рака желудка (GC) [1]. Инфекция остается скрытым у большинства инфицированных пациентов, и лишь меньшинство людей с H. Pylori
инфекции когда-либо развития заболевания [3]. Uemura и др. сообщил, что GC, разработанный в приблизительно 3% H. пилори
-infected пациентов в течение периода наблюдения 10 лет, по сравнению с ни один из неинфицированных больных [4]. В дополнение к принимающей, окружающей среды, а также диетические факторы, различия в вирулентности H. штаммы пилори
связаны с различными результатами H. пилори
инфекции. Факторов вирулентности H. пилори
, такие как CaGa, Вака, DUPA, ICEA, oipA,
и Baba
, было показано, что предсказатели тяжелых клинических исходов [5], [6]. Важно отметить, что большинство из этих факторов вирулентности, связаны друг с другом; CaGa
-положительные штаммы также обладают Вака
s1 /m1 тип и они далее тесно связаны с наличием Baba
и oipA
"на" статус [5].
<р> генетическое разнообразие внутри H. пилори
больше, чем у большинства других бактерий [7], и примерно в 50 раз больше, чем у человеческой популяции [8]. Кроме того, частая рекомбинация между различными H. штаммы пилори
[9] приводит лишь частичной неравновесия по сцеплению между полиморфных локусов, что обеспечивает дополнительную информацию для населения генетического анализа [10]. В последнее время геномная разнообразие внутри H. Pylori
популяции была рассмотрена полилокусной последовательность ввода метода (MLST) с использованием семи генов домашнего хозяйства ( ATPA, EFP, MutY, PPA, trpC, Urei,
и yphC
) [10] - [12]. В настоящее время семь типов населения были определены на основе географических ассоциаций и обозначается следующим образом: hpEurope, hpEastAsia, hpAfrica1, hpAfrica2, hpAsia2, hpNEAfrica и hpSahul [10] - [12]. hpEastAsia является общим в H. пилори
изолятов из Восточной Азии, и их можно разделить на три подгруппы hspEAsia, hspAmerind и hspMaori. hpEurope включает в себя почти все H. штаммы пилори
изолированы от этнических европейцев, в том числе людей из стран, колонизированных европейцами. H. пилори
предсказывается, распространилась из Восточной Африки за тот же период времени, как анатомически современных людей (~58,000 лет назад), так и остался тесно связан с их человеческим хозяевам с тех пор [5], [11], [12] .
<р> возраст стандартизированный показатель заболеваемости (ASR) ГК в Колумбии, как сообщается, быть относительно высоким (13,4 /100 000 населения), по сравнению с другими странами Южной Америки (в среднем ASR = 10,3 /100 000 населения) (Международное агентство по Исследования рака; GLOBOCAN2012, http://globocan.iarc.fr/). Следует отметить, что ГК является более распространенным в колумбийской горной местности, чем на побережье [13], [14]. де Сабле и др. проводили MLST для определения филогеографического различия между горных и прибрежных районах [15]. Интересно, что они обнаружили, что все CaGa
-положительные штаммы из регионов повышенного риска GC принадлежали hpEurope, тогда как hpEurope и hpAfrica1 сосуществуют в регионах GC с низким уровнем риска [15]. Кроме того, субъекты инфицированы hpEurope штаммов H. пилори
показали более высокие гистологической оценки, чем у инфицированных штаммов hpAfrica1. Эти наблюдения указывают на то, что филогеографического происхождение определяется MLST может быть использован как предсказатель GC риска.
<Р> Тем не менее, эти исследования не рассматривали филогенетическое происхождение в соответствии с клиническими результатами, и, таким образом, остается неясным, является ли филогенетического происхождения действительно связаны с клиническими результатами в Колумбии. В данном исследовании мы рассмотрели филогенетическое происхождение H. Pylori
штаммы из GC и двенадцатиперстной кишки (DU) пациентов, живущих в Боготе, столице и самом большом городе, расположенном в регионе Анд в Колумбии.
были получены из слизистой оболочки желудка H. Pylori
-infected колумбийские пациентов с GC и DU, которые прошли эндоскопию в Universidad Nacional де Колумбии, Богота, Колумбия. GC и DU были идентифицированы с помощью эндоскопии, и GC было дополнительно подтверждено гистопатологией [16]. У больных с историей частичной резекции желудка были исключены. Письменное информированное согласие было получено от всех участников, и протокол был одобрен Комитетом по этике Национального университета Колумбии.
Выделение и генотипирование H. пилори
<р> Образцы антральных биопсии были получены для выделения H. пилори
с использованием стандартных методов культивирования, как описано ранее [17]. H. пилори
ДНК экстрагировали из культур сливающийся плит вспененных из одной колонии с использованием коммерчески доступного набора (Qiagen, Valencia, CA). Статус CaGa
определяли с помощью полимеразной цепной реакции (ПЦР) для сохраняющегося области CaGa
и для прямого секвенирования с использованием пары праймеров cagTF; 5'-АСС СТА GTC GGT AAT GGG-3 'и cagTR; 5'-GCT TTA GCT TCT GAY НКД GC-3 '(Y = C или T), сконструированные в 3' области повтора CaGa
, как описано ранее [18]. Условия ПЦР были начальная денатурация в течение 5 мин при 95 ° С, 35 шагов амплификации (95 ° C в течение 30 сек, 56 ° C в течение 30 с, и 72 ° C в течение 30 с), и конечным удлинением цикла 7 мин при 72 ° С, используя смесь Taq-ДНК-полимеразы (TOYOBO, Осака, Япония). Отсутствие CaGa
было подтверждено наличием CaGa
пустой сайт, как описано ранее [19]. ПЦР-продукты очищали с QIAquick Purification Kit (QIAGEN) в соответствии с инструкциями изготовителя. Амплифицированный фрагмент был обнаружен с помощью электрофореза в агарозном геле 1,5%, который был впоследствии окрашивали бромистым этидием и визуализировали с использованием ультрафиолетового транс-осветителя.
<Р> вача
генотипирование (s1, s2, м1 и м2) проводили, как описано ранее [20], [21]. Грунтовки для сигнальной области получали фрагмент длиной 259 пар оснований и 286 пар оснований для S1 и S2 вариантов, соответственно. Грунтовки для средней области получали фрагмент длиной 570 пар оснований и 645 пар оснований для m1 и m2 вариантов соответственно.
филогенетический анализ H. Pylori
штаммы
<р> MLST генов семи домашнего хозяйства ( ATPA, EFP, MutY, PPA, trpC, Urei, yphC
) определяли с помощью ПЦР на основе секвенирования, как описано ранее [7 ], [22]. Прямое секвенирование ДНК проводили с использованием AB 3130 Genetic Analyzer (Applied Biosystems, Foster City, CA) в соответствии с инструкциями изготовителя. Для построения филогенетического дерева на основе MLST генотипов, последовательности наборов данных из 7 домашнего хозяйства генов hpAfrica2, hpAfrica1, hpNEAfrica, hpEurope, hpSahul, hpAsia2, hspMaori, hspAmerind и hspEAsia штаммов (60, 181, 61, 566, 49, 17 , 80, 18, и 177 штаммов, соответственно, 1,209 штаммы в общей сложности) были получены из базы данных PubMLST (http://pubmlst.org/). Эти наборы данных последовательности были объединены с нашими данными из 66 колумбийских штаммов. Сосед присоединяющиеся деревья были построены с использованием MEGA 5.0 Кимура-2 параметра [23].
Структура населения Анализ H. штаммы пилори
<р> Мы проанализировали структуру бактериальной популяции с использованием программного обеспечения (СТРУКТУРА v.2.3.2) [24]. Цепи Маркова Монте-Карло (MCMC) моделирования структуры были проведены в модели смеси с выжигания 20000, а затем 30000 итераций для каждого прогона. Число предварительных популяций (K) была установлена от 7 до 10 и 5 трасс были выполнены для каждого К.
<р> Мы включили 35 пациентов GC и 31 пациентов DU. Штаммы, выделенные от больных GC включены 24 CaGa
-положительным и 11 CaGa
-отрицательное. Двадцать девять были Вака
s1m1 генотип и 6 были Вака
s2m2 генотип. Все 24 CaGa
-положительные штаммы GC показал Вака
s1m1 генотип. Для 31 штаммов от больных DU, 22 штаммы были CaGa
-положительным, а остальные 9 были CaGa
-отрицательное. Вака
s1m1 генотип был обнаружен у 16 штаммов, Вака
s1m2 в 5 штаммов и Вака
s2m2 в 10 штаммов. Среди 22 CaGa
-положительные штаммы из пациентов DU, 14 штаммов обладали Вака
s1m1 генотип. Пять штаммов были s1m2, а остальные 3 штамма были s2m2.
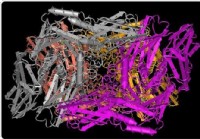
<Р> Типы популяций 35 штаммов, выделенных от больных с GC и 31 из ОУ были проанализированы с помощью MLST. Филогенетическое древо из 66 колумбийских штаммов на основе последовательностей MLST и типов заболевания показаны на рисунке 1. GC и штаммы DU были разбросаны в дереве MLST; Таким образом, мы не обнаружили никакой разницы в филогенетического распределения между GC и штаммов DU. Рисунок 2А показывает CaGa
статус штаммов на дереве MLST. Штаммы от CaGa
-положительным и CaGa
-отрицательное не могли быть разделены четко, хотя и CaGa
-отрицательные штаммы были относительно сгруппированы в той же отрасли. Фигура 2B показывает Вака
типы штаммов на том же дереве MLST. Шестнадцать Вака
s2m2 штаммы были сгруппированы в подотрасли вместе с двумя s1m2 и шестью s1m1 штаммов. Остальные три Вака
s1m2 штаммы были расположены среди Вака
s1m1 штаммов 39.
<Р> Далее, мы построили филогенетическое дерево на основе MLST последовательностей 66 штаммов колумбийских и 1,209 эталонные штаммы hpAfrica2, hpAfrica1, hpNEAfrica, hpEurope, hpSahul, hpAsia2, hspMaori, hspAmerind и hspEAsia, депонированные в базе данных pubMLST (60, 181, 61, 566, 49, 17, 80, 18, и 177 штаммов, соответственно) ( Рисунок 3). Среди 66 колумбийских штаммов, только один CaGa
-положительным штамм был расположенный среди подотраслей hpAfrica1. Оставшиеся 65 штаммов были рассеяны среди hpEurope подотраслей независимо от видов заболеваний. Таким образом, не было обнаружено никакой связи между ветвление филогенетического древа и клинические исходы.
<Р> Для исследования структуры популяции колумбийских штаммов, мы провели анализ населения с помощью Структура программного обеспечения [24]. Для этого анализа мы использовали те же штаммы 1257 (1209 эталонных штаммов и 66 колумбийских штаммы), которые были использованы для филогенетического анализа MLST. Структура программного обеспечения выполняет MCMC моделирования для классификации отдельных лиц для заданного числа популяций (K). Для данного K, структура определяет компоненты популяции K и представляет их K цветов, используя один цвет для представления одного компонента населения. Мы провели анализ структуры путем установки K от 7 до 10 и выполненных моделирования в пять раз для каждого К. На рисунке 4 показаны результаты K = 9 и 10, чья задняя вероятность является лучшим из пяти трасс (наиболее вероятные результаты). Каждая вертикальная линия гистограммы представляет один штамм и цвета линии указывают на популяции, к которым штамм может принадлежать. Длины цвета в линии пропорциональны вероятности того, что штамм принадлежит к каждой популяции. Поскольку группа hpEurope примесь нескольких происхождения, эта группа показывает сложные цвета.
<Р> В результате K = 7, колумбийские штаммы показали общие цвета с hpEurope штаммов (рис 4A). Когда K был установлен в 10, колумбийские и несколько штаммов hpEurope представил различный цвет от других, который представляет собой компонент популяции, специфичные для этих штаммов (светло-зеленые полосы, отмеченные звездами на рисунке 4б). Это означает, что эти штаммы имеют принципиальное сходство с европейскими штаммов, но отличаются от других, если детально рассмотрены. Рисунок 4C является увеличение колумбийского штаммов. Стержни выровнены слева направо в порядке уменьшения светло-зеленого компонента убывания. CaGa
типы и заболевания показаны знаками ниже гистограммы. Как видно из этого рисунка видно, что CaGa
-отрицательные штаммы были частыми на правом торце. В то время как большинство колумбийских штаммов имели конкретный компонент и /или общий компонент населения с hpEurope, один штамм показал более высокое сходство с hpAfrica1 (фиг.4С, бар помеченный треугольником). Этот штамм соответствует тому, который принадлежит к hpAfrica1 подветви в филогенетического дерева MLST (рисунок 3).
<Р> В данных, взятых из PubMLST, мы выбрали 63 hpEurope штаммы, которые содержали светло-зеленый компонент населения в 10% или выше, и исследовали их местоположение выборки. Двадцать пять штаммов были выделены из Колумбии, хотя они были классифицированы как hpEurope, 19 были из Испании, 4 были из Венесуэлы, а другие из разных стран (таблица S1).
Обсуждение
<р> хост , экологические и диетические факторы, а также различия в H. штаммы пилори
связаны с различными результатами H. пилори
инфекции. В целом, распределение заболеваемости GC тесно связана с этим H. Pylori
групп определяется с помощью анализа населения на основе MLST [25]. Высокая частота GC была обнаружена в тех регионах, где преобладают штаммы hpEastAsia (особенно hspEAsia). Тем не менее, частота GC является очень низким в Африке, где большинство штаммов hpNEAfrica, hpAfrica1 или hpAfrica2, и в Южной Азии, где большинство штаммов hpAsia2. В целом, низкая заболеваемость раком желудка в странах Африки и Южной Азии можно было бы объяснить, по крайней мере частично, различными генотипами H. пилори
циркулирующих в различных географических районах. Интересно, что недавний доклад о CaGa
-положительные штаммы в Колумбии показали, что все штаммы из регионов повышенного риска ГК принадлежат hpEurope, тогда как hpEurope и hpAfrica1 штаммы сосуществуют в регионах с низким уровнем риска [15]. Кроме того, субъекты инфицированы hpEurope штаммов H. пилори
показали более высокие гистологической оценки, чем у инфицированных штаммов hpAfrica1. Авторы пришли к выводу, что разница в популяциях бактерий может быть использован как предсказатель GC риска.
изолированы от колумбийских пациентов с GC и DU, но не гастритом. У пациентов с гастритом только на момент эндоскопии может развиться DU и GC позже в жизни; И наоборот, пациенты DU редко развиваются GC в своей жизни [4], [26]. Поэтому, хотя <ЕМ> Н. Pylori
инфекция способствует развитию как GC и DU, GC и DU рассматриваются результаты варианта. В данном исследовании мы не обнаружили никакой разницы в филогенетических групп между GC и штаммов DU. были найдены Наши колумбийские штаммы принадлежат к hpEurope, за исключением одного штамма hpAfrica1 исключением. Хотя колумбийские штаммы были разделены на несколько подотраслей в филогенетических деревьев, не было никакой корреляции между ветвями и болезнями (рисунки 1 и 3). Таким образом, топология дерева MLST не действует в качестве маркера для оценки ГХ и DU риск для штаммов hpEurope в Колумбии. На самом деле, даже в исследовании, проведенном де Сабле и др., Все CaGa
-отрицательные штаммы также считаются принадлежащими к hpEurope [15]. Авторы утверждают, что субъекты, инфицированные CaGa
-отрицательные штаммы были очень низкие гистологической оценки; Поэтому, hpEurope штаммы без присутствия CaGa
может быть менее вирулентными. Кроме того, мы ранее сообщали, что штаммы, имеющие более трех повторных областей региона 3 'в CaGa
были связаны со значительно более высокие оценки для слизистой оболочки желудка атрофии и кишечной метаплазии, чем те, с меньшим количеством повторных областей в Колумбии [20 ]. Распространенность штаммов с более трех повторных областей была выше в GC, чем DU [20]. Таким образом, тип CaGa
а не филогеографического происхождения может быть лучшим предсказателем GC для штаммов hpEurope [27].
<Р> Ранее мы обнаружили, что хотя OipA и CAG
PAI связаны между собой, только OipA был независимым фактором риска для DU [28]. OipA в CaGa
-положительные штаммы могут способствовать филогеографического вариации определяется с помощью анализа MLST. Кроме того, мы сообщали, что jhp0045
и jhp0046
были связаны с GC в разделе CaGa
-положительные штаммы в Колумбии [29]. Кроме того, мы сообщали, что инфекции с DUPA
-положительные штаммы повышает риск для DU, но были защитными против GC в Колумбии [30]. Таким образом, состояние других факторов вирулентности могут быть соотнесены с филогенетического дерева, полученного выполнением MLST [5]. Отношения между филогении генов домашнего хозяйства и CAG
PAI филогении сообщалось [31]. Филогения наиболее CAG
генов PAI был похож на MLST, указав, что CAG
PAI, вероятно, приобрел только один раз H. пилори
, и ее генетическое разнообразие отражает изоляцию на расстоянии, которое сформировало это виды бактерий, так как современные люди мигрировали из Африки [31]. В настоящем исследовании, филогенетическое дерево, основанное на MLST не коррелировала с типом CaGa
или Вака
, хотя штаммы с CaGa
-отрицательное или Вака
s2m2 генотипы были сгруппированы относительно. Только один штамм принадлежал hpAfrica1, и этот штамм обладал CaGa
, предполагая, что CaGa
и Вака
генотипы были не определяющими факторами для филогенетического происхождения на основе MLST в Андах регион в Колумбии. Тем не менее, мы уже сообщали о том, что тип CaGa
был связан с филогенетической кластера в Окинаве, Япония [22], [27]. Штаммы из Окинавы были разделены на 3 субпопуляции и одной примеси группы под влиянием западных штаммов. Филогеографического топологии и субпопуляции были связаны с клиническими результатами; Однако, разница в филогеографического топологии было связано со статусом CaGa
(типа восточноазиатской, западного типа CaGa
и CaGa
-отрицательное) и Вака
(m1 или m2) из субпопуляций. GC был более распространенным в кластере, содержащем в основном восточноазиатского типа CaGa
штаммов. Результаты MLST показали, что распространенность GC в каждой группе не отличались, когда только западного типа штаммы CagA
или CaGa
-отрицательные штаммы были использованы [27]. Таким образом, филогеографического происхождения может выступать в качестве сбивающий фактор в прогнозировании факторов вирулентности, но не исход заболевания (например, в hspEAsia, большинство из них CaGa
-положительным, Вака
s1 /m1 тип, Baba
-положительным и oipA
"на" состоянии). В недавнем исследовании, не только H. Pylori
родословной, но и коэволюция между человеком и H. пилори
оказался лучшим предсказателем для предраковых поражений [32]. Необходимы дальнейшие исследования, чтобы подтвердить связь между родовым происхождением H. пилори
независимо от факторов вирулентности и клинических исходов.
<р> В этом исследовании мы включили пациентов метисов в основном проживающих в городе Богота, который находится в горной местности (2600 м над уровнем моря). Эта область обычно заселены метисов, смешанной популяции, которые произошли от коренных американцев (Америнды) и европейских народов, начиная от испанского колониального периода [33]. Америнды предположили, были инфицированы штаммами первоначально hspAmerind, которые принадлежат к субпопуляции hpEastAsia [10]. Таким образом, мы ожидали, что H. пилори
изолированы от метисов будет отображать смешанный тип hpEurope и hspAmerind. Интересно, что во всех случаях, кроме одного, в нашем исследовании были инфицированы hpEurope штаммов, в соответствии с предыдущим исследованием [15]. Структурный анализ показал, что некоторые штаммы с hpEurope показали мозаичность; Тем не менее, они не представляли собой смесь hpEurope и hspAmerind. Это наблюдение подтверждает гипотезу о том, что штаммы hpEurope обладают конкурентным преимуществом перед штаммов коренных hspAmerind, как описано ранее [34], [35]. Удивительно, но наше предыдущее исследование показало, что даже в 16 штаммов, выделенных из Huitoto, примитивной, изолированной группы, живущей в амазонских джунглях в Колумбии, 4 штаммы были инфицированы hspAmerind, в то время как остальные 12 были hpEurope штаммы [12]. Кроме того, мы обнаружили, что некоторые индейские штаммы из Колумбии обладали западного типа CaGa
но не восточно-азиатского типа CaGa
[36]. Остается неясным, почему большинство индейцах H. штаммы пилори
имеют генотипы типичных штаммов из западных стран, даже в местах, где западное влияние, как представляется, было минимальным. hpEurope, а не штаммы hspAmerind, может быть легко адаптирована к условиям окружающей среды и селективного давления, оказываемого хозяина. Две гипотезы, одна на основе конкуренции деформации и другие деформации на подрывной деятельности путем трансформации, были предложены в качестве причины перемещения hspAmerind по hpEurope [34]. Дальнейшее исследование будет необходимо определить механизм (ы), отвечающий за В естественных условиях
потеря америндами штаммов, когда в условиях конкуренции с штаммами Старого Света. Интересно, что мы обнаружили, что некоторые колумбийские штаммы показали определенный компонент населения, и этот компонент поделили не только с другими южноамериканскими штаммов, но и с испанских штаммов, депонированных в PubMLST. Город Богота, основанный в 1538 году, стал одним из главных административных центров испанских владений в Новом Свете (наряду с Лимой и Мехико) [33]. Наш анализ показывает, что hpEurope штаммы в Колумбии были введены в основном испанских иммигрантов. Население типирование H. пилори
является полезным инструментом для картирования миграционных моделей человека [37]; В самом деле, наши результаты могут быть полезны для выяснения тех, у современных людей в Южной Америке. Хотя наш анализ MLST на основе семи генов домашнего хозяйства предполагает, что большинство колумбийских штаммов были заменены штаммов hpEurope, мощи предков аборигенных штаммов могут оставаться в других частях генома. Более обширные во всем мире и геномные исследования помогут нам глубже понять эволюцию и структуру населения H. пилори
.
<р> Одним из потенциальных ограничений нашего исследования стоит отметить, что мы не смогли получить достаточную информацию о этнической принадлежности пациентов, от которых H. штаммы пилори
были изолированы. В самом деле, было высказано предположение, что хозяин происхождения может повлиять на клинические результаты, как описано выше [32]. Например, воспалительных полиморфизма генов цитокинов ( IL-1
кластера генов, TNF-α,
IL-10
и IL-8
) сообщалось, что коррелирует с GC [38] - [43]. Кроме того, история семьи GC было показано, внести свой вклад в клинические исходы [44]. В дополнение к другим H. Pylori
факторы вирулентности, эта информация может быть полезна для различения между GC и DU риска. Дальнейшие исследования должны выяснить связь между H. Pylori
генотипов и исходы заболевания.
<р> Наше исследование показало, что филогеографического происхождение от MLST не было достаточно, чтобы различать между GC и DU риска в регионе Анд в Колумбии. Филогенетический анализ, включая факторов вирулентности H. пилори
может быть необходимо более точно определить клинические исходы. Кроме того, наш анализ также показывает, что hpEurope штаммы в Колумбии были введены в первую очередь испанских иммигрантов и их генотип не находился под влиянием америндами.
Поддержка Информация
таблице S1.
DOI: 10.1371 /journal.pone.0105392.s001
(DOCX)
<р> Мы благодарим г-жу Миюки Мацуда за отличную техническую помощь
.
 Нездоровый микробиом кишечника снижает синаптическую обрезку мозга,
ухудшает обучение Новое исследование Weill Cornell Medicine и Корнельского университета дало больше объяснений того, как кишечные микробы взаимодействуют с нейронами мозга. Изучение, опубликовано в жу
Нездоровый микробиом кишечника снижает синаптическую обрезку мозга,
ухудшает обучение Новое исследование Weill Cornell Medicine и Корнельского университета дало больше объяснений того, как кишечные микробы взаимодействуют с нейронами мозга. Изучение, опубликовано в жу
 Новое исследование может помочь предотвратить смертельные инфекции у младенцев
Недоношенные дети, рожденные до 28-30 недель жизни, подвержены высокому риску многих осложнений, среди которых очень велики шансы умереть от инфекции, начинающейся в кишечнике. Новое исследование, опу
Новое исследование может помочь предотвратить смертельные инфекции у младенцев
Недоношенные дети, рожденные до 28-30 недель жизни, подвержены высокому риску многих осложнений, среди которых очень велики шансы умереть от инфекции, начинающейся в кишечнике. Новое исследование, опу
 Тиопурины могут помочь остановить репликацию вируса в коронавирусах человека
Исследователи из отдела микробиологии и иммунологии, Университет Далхаузи, Университет Калгари, и кафедра биохимии и молекулярной биологии, Университет Британской Колумбии, Канада, работал с человечес
Тиопурины могут помочь остановить репликацию вируса в коронавирусах человека
Исследователи из отдела микробиологии и иммунологии, Университет Далхаузи, Университет Калгари, и кафедра биохимии и молекулярной биологии, Университет Британской Колумбии, Канада, работал с человечес