Pozadie
Nedávna správa ukázala, že fylogenetický pôvod Helicobacter pylori sme zahrnuli 35 pacientov s rakovinou žalúdka a 31 pacientov s dvanástnikovým vredom, ktoré sú považované za výsledky varianty. Genotypy ČAGA stroje a krava H. pylori Závery Naša štúdia ukázala, že phylogeographic pôvod určí MLST bol nedostatočný pre rozlišovanie medzi rakovinou žalúdka a dvanástnika riziko vredu medzi hpEurope kmene v andskom regióne v Kolumbii. Naša analýza tiež naznačuje, že hpEurope kmene v Kolumbii boli primárne zavedené španielskych prisťahovalcoch Citácia :. Shiota S, R Suzuki, Matsuo Y, Miftahussurur M, Tran TTH, Binh TT, et al. (2014) Helicobacter pylori Editor: Masaru Katoh, National Cancer Center, Japonsko prijatá: 7. marca 2014; Prijaté: 23.júla 2014; Uverejnené: 14. augusta 2014 Copyright: © 2014 Shiota et al. Toto je článok o otvorený prístup distribuovaný pod podmienkami Creative Commons Attribution licencie, ktorá umožňuje neobmedzené použitie, distribúciu a reprodukciu v nejakom médiu, za predpokladu, že pôvodný autor a zdroj sú pripísané Dostupné dát: Text. autori potvrdzujú, že všetky údaje súvisiace závery sú plne k dispozícii bez obmedzenia. Nukleotidov sekvenčné dáta Uvádzané sú k dispozícii pod DDBJ prístupové čísla AB923031 k AB923492 Financovanie :. Táto správa je založená na prácu podporovaný z časti dotácie z National Institutes of Health (DK62813) (RR), a granty -v-Aid pre vedecký výskum od Ministerstva školstva, kultúry, športu, vedy a technológie (MEXT) Japonska (22390085, 22659087, 24406015 a 24659200) (YY), (23790798) (SS) a zvláštnym spôsobom koordinované fondy pre podporu Veda a technológie z Japonska Science and Technology Agency (JST). Platcovia mal žiadnu úlohu v dizajne štúdie, zber a analýzu dát, rozhodnutie publikovať, alebo prípravu rukopisu Konkurenčné záujmy: .. Autori vyhlásili, že žiadne konkurenčné záujmy neexistujú Úvod Helicobacter pylori genetická diverzita vnútri H. pylori vek štandardizovaná miera incidencie (ASR) GC v Kolumbii je hlásený byť pomerne vysoká (13,4 /100 tisíc obyvateľov) v porovnaní s ďalšími krajinami južnej Ameriky (priemerný ASR = 10,3 /100 tisíc obyvateľov) (Medzinárodná agentúra pre výskum rakoviny, GLOBOCAN2012, http://globocan.iarc.fr/). Pozoruhodne, GC je častejšia u kolumbijskej horskej oblasti, než na pobreží [13], [14]. de Sablet et al. vykonané MLST určiť phylogeographic rozdiely medzi horské a pobrežné regióny [15]. Zaujímavé je, že zistili, že všetci ČAGA Tieto štúdie neskúmal fylogenetický pôvod podľa klinické výsledky, a tak zostáva nejasné, či fylogenetického pôvodu je skutočne spojená s klinickými výsledkami v Kolumbii. V tejto štúdii sme skúmali fylogenetický pôvod H. pylori Materiály a metódy Pacienti H. pylori vzorky antrálnej biopsie boli získané pre izoláciu H. pylori krava MLST z domácnosti génov siedmich ( ATPA, EFP, mutya, PPA, trpko, UREI, yphC Analyzovali sme bakteriálnej štruktúru obyvateľstva pomocou softvéru štruktúry (v.2.3.2) [24]. Markov Chain Monte Carlo (MCMC) simulácia štruktúry boli prevádzkované v prímesou modelu s vypálenie 20.000, nasledovalo 30.000 iterácií pre každého behu. Počet predbežných populácií (K) bola stanovená od 7 do 10 a 5 behy boli vykonané pre každú K. nukleotidovej sekvencie nukleotidov sekvenčné dáta Uvádzané sú k dispozícii pod DDBJ číslami AB923031 na AB923492. Výsledky sme zahrnuli 35 pacientov GC a 31 pacientov DU. Kmene izolované od pacientov GC zahrnuté 24 ČAGA Typy Populačný 35 kmeňov izolovaných od pacientov s GC a 31 od spoločnosti Du boli analyzované pomocou MLST. Fylogenetický strom 66 kolumbijskej kmeňov na základe sekvencií MLST a typy ochorenia sú uvedené na obrázku 1. GC a kmene DU boli rozptýlené vo stromu MLST; Preto sme nenašli žiadny rozdiel v fylogeneticky rozdelenie medzi GC a kmene DU. Obrázok 2A ukazuje ČAGA Ďalej sme skonštruovali fylogenetický strom založený na MLST sekvencií 66 kolumbijských kmeňov a 1,209 referenčné kmene hpAfrica2, hpAfrica1, hpNEAfrica, hpEurope, hpSahul, hpAsia2, hspMaori, hspAmerind a hspEAsia, uložené na pubMLST databázu (60, 181, 61, 566, 49, 17, 80, 18, a 177 kmeňov, v uvedenom poradí) ( obrázok 3). Medzi kolumbijskej kmeňov 66, iba jeden ČAGA Ak chcete skúmať štruktúru obyvateľstva kolumbijskej kmeňov, sme vykonali analýzu populácie pomocou Structure [24]. Pre túto analýzu sme použili rovnaké 1,257 kmene (1,209 referenčné kmene a 66 kolumbijských kmene), ktoré boli použité pre fylogeneticky analýzy MLST. ŠTRUKTÚRA softvér vykonáva MCMC simulácie pre klasifikáciu jednotlivcov pre daný počet populácií (K). Pre danú K, štruktúry určuje zložiek K obyvateľstva a predstavuje im K farby s použitím jednej farby reprezentovať jednu zložku obyvateľstva. Vykonali sme štruktúrne analýza nastavením K od 7 do 10 a vykonala simulácie päťkrát pre každý K. Obrázok 4 ukazuje výsledky K = 9 a 10, ktorých neskoršie pravdepodobnosti je najlepší z piatich pokusov (najpravdepodobnejší výsledky). Každá zvislá čiara zo stĺpcových grafov predstavuje jeden kmeň a farby čiary ukazujú populácie, ktoré môže patriť kmeň. Dĺžky farby v rade sú proporcionálne k pravdepodobnosti, že kmeň patrí ku každej populácii. Vzhľadom k tomu, hpEurope skupina je prímes niekoľkých pôvodov, táto skupina vykazuje zložité farby. V dôsledku K = 7, kolumbijskej kmene vykazovali spoločné farby s hpEurope kmeňmi (obrázok 4A). Keď bola nastavená na 10 K, kolumbijskej a niekoľko kmeňov hpEurope predstavil odlišnú farbu od ostatných, ktoré predstavuje súčasť populácie špecifickú pre tieto kmene (svetlo zelené koridory označené hviezdičkou na obrázku 4B). To znamená, že tieto kmene majú základné podobnosť s európskym kmeňom, ale sa odlišujú od ostatných, ak podrobne preskúmaná. Obrázok 4C je zväčšenie kolumbijských kmeňov. Tyče boli zarovnané zľava doprava, v zostupnom poradí podľa svetlo zelenej zložky. ČAGA Vo údajov zisťovaných z PubMLST, sme vybrali 63 hpEurope kmeňov, ktoré obsahovali svetlo zelenej populácie zložku na 10% alebo vyššia, a skúmal ich umiestnenia vzorkovania. Dvadsaťpäť kmene boli izolované z Kolumbie, hoci boli klasifikované ako hpEurope, 19 bolo zo Španielska, 4 boli z Venezuely, a iní sú z rôznych krajín (pozri tabuľku S1). hostiteľských , životného prostredia a diétne faktory a rozdiely v H. Kmene pylori V tejto štúdii sme zahrnuli H. pylori Už skôr sme zistili, že hoci OipA a CAG V tejto štúdii sme zaradili pacientov mestici žijúci prevažne v meste Bogota, ktorá sa nachádza v horskej oblasti (2600 m nm). Táto oblasť je všeobecne obývaný mesticov, zmiešané populáciu, ktorí zostúpili z domorodých Američanov (Amerindians) a Európskych ľudí z doby španielskej kolonizácie obdobie [33]. Amerindians existujú hypotézy, že boli nakazení pôvodne s kmeňmi hspAmerind, ktoré patria k podskupine hpEastAsia [10]. Preto sme očakávali, že H. pylori Jednou z potenciálnych obmedzení našej štúdie Za zmienku stojí, že sme nemohli získať dostatok informácií o etnicite pacientov od koho H. Kmene pylori Záver Naša štúdia ukázala, že phylogeographic pôvod podľa MLST nebol dostatočný rozlišovať medzi GC a rizikom DU v andskom regióne v Kolumbii. Fylogeneticky analýzy, vrátane faktorov virulencie zo H. pylori Poďakovanie Ďakujeme pani Miyuki Matsuda za vynikajúcu technickú pomoc
založený na multi-locus sekvenčná typizácia (MLST) bola významne spojená s závažnosť gastritídy v Kolumbii. Avšak, potenciálny vzťah medzi fylogenetického pôvodu a klinickým výsledkom nebola skúmaná v tejto štúdii. V prípade, že fylogeneticky pôvodu, skôr než virulentné faktory boli skutočne spojená s klinickými výsledkami, identifikácia populácie s vysokým rizikom rakoviny žalúdka v Kolumbii by byť pomerne jednoduché. V tejto štúdii sme skúmali fylogenetický pôvod kmeňov od rakovinou žalúdka a pacientov s dvanástnikovým vredom žijúcich v Bogote, Kolumbii.
Metódy
boli stanovené pomocou PCR. Genealógia týchto kolumbijskej kmeňov bola analyzovaná MLST. Bakteriálna populačná štruktúra bola analyzovaná pomocou Structure.
Výsledky
kmene od rakovinou žalúdka a pacientov s dvanástnikovým vredom boli rozptýlené v fylogenetického stromu; Preto sme sa nezistil žiadny rozdiel v fylogeneticky rozdelení medzi rakovinou žalúdka a kmeňov s dvanástnikovým vredom v skupine hpEurope v Kolumbii. Šesťdesiat šesť kmene, až na jednu výnimku, bolo klasifikované ako hpEurope bez ohľadu na ČAGA stroje a Vaca
genotypov a typu ochorenia. štruktúrne analýza odhalila, že kolumbijské kmene hpEurope majú fylogeneticky pripojenie k španielskym kmeňom.
z rakoviny žalúdka a dvanástnikové vredy Show Same Phylogeographic pôvodu v andskom regióne v Kolumbii. PLoS ONE 9 (8): e105392. doi: 10,1371 /journal.pone.0105392
je špirálová gramnegatívne baktérie, ktorá infikuje viac ako polovica svetovej populácie [1]. Transmisný mechanizmus H. pylori
nebol ešte úplne objasnené, ale z človeka na človeka šírenie prostredníctvom ústnych-ústne alebo fekálne-orálny trasách je považovaný za najpravdepodobnejší [2]. H. pylori
infekcie je teraz prijímaná, ktoré majú byť spojené s vážnymi chorobami gastritídou spojené, vrátane žalúdočného vredu a rakoviny žalúdka (GC) [1]. Infekcia zostáva latentný u väčšiny infikovaných pacientov, a len menšina jedincov s H. pylori
infekcie niekedy vyvinúť ochorenie [3]. Uemura et al. hlásené, že GC vyvinuté v približne 3% H. pylori
-infected pacientov počas pozorovací obdobie 10 rokov, v porovnaní s žiadny z neinfikovaných pacientov [4]. Okrem hostiť, životného prostredia a diétne faktory, rozdiely vo virulenciu H. Kmene pylori
sú spojené s rôznou výsledky H. pylori
infekciu. Virulentné faktory H. pylori
, ako je napríklad ČAGA, krava, Dupa, ICEA, oipA, stroje a Baba
, bolo preukázané, že prediktory závažné klinické dôsledky [5], [6]. Dôležité je, že väčšina z týchto faktorov virulencie sú spojené medzi sebou; ČAGA
-pozitívnych kmene tiež mať Vaca
S1 /M1 typu a sú ďalej úzko súvisí s prítomnosťou Baba stroje a oipA
"on" stave [5].
je väčší než u väčšiny ostatných baktérií [7], a asi 50-krát vyššie ako u ľudskej populácie [8]. Okrem toho časté rekombinácie medzi rôznymi H. pylori
kmene [9] vedie len k čiastočnej väzobnej nerovnováhe medzi polymorfné loci, ktorý poskytuje dodatočné informácie pre populačnej genetickej analýzy [10]. V poslednej dobe, genomické rozmanitosť vo H. pylori
populácia bola skúmaná pomocou multi-locus sekvenčná typizácia metódou (MLST) pomocou siedmich upratovanie gény ( ATPA, EFP, mutya, PPA, trpko, UREI, stroje a yphC
) [10] - [12]. V súčasnej dobe už sedem typov populácie boli identifikované na základe zemepisných asociácií a označujú takto: hpEurope, hpEastAsia, hpAfrica1, hpAfrica2, hpAsia2, hpNEAfrica a hpSahul [10] - [12]. hpEastAsia je bežné v H. pylori
izoluje z východnej Ázie, a môžu byť rozdelené do troch podskupín hspEAsia, hspAmerind a hspMaori. hpEurope zahŕňa takmer všetky H. pylori
kmene izolované z etnických Európanov, vrátane ľudí z krajín kolonizovaných Európanmi. H. pylori
Predpokladá sa šíri z východnej Afriky v rovnakom časovom období ako anatomicky moderných ľudí (pred ~ 58,000 rokov), a zostal úzko spojené so svojimi ľudskými hostiteľmi odvtedy [5], [11], [12] .
-pozitívnych kmeňov z GC vysoko rizikových oblastiach patril hpEurope, zatiaľ čo hpEurope a hpAfrica1 koexistovať v GC s nízkym rizikom regiónoch [15]. Navyše, predmety infikované hpEurope kmene H. pylori
vykazovali vyššie histopatologické skóre ako nakazených kmene hpAfrica1. Tieto pozorovania naznačujú, že phylogeographic pôvod určený MLST môže byť použitá ako prediktor rizika GC.
kmene z GC a dvanástnikové vredy (DU) pacientov žijúcich v Bogote, hlavným a najväčším mestom sa nachádza v andskom regióne v Kolumbii.
kmene boli získané z žalúdočnej sliznice H. pylori
-infected kolumbijskej pacienti s GC a DU, ktorí podstúpili endoskopiu na Universidad Nacional de Colombia, Bogotá, Kolumbia. GC a DU boli identifikované pomocou endoskopie a GC bola ďalej potvrdená histopatológie [16]. Pacienti s anamnézou parciálny resekcii žalúdka boli vylúčené. Písomný informovaný súhlas bol získaný od všetkých účastníkov, a protokol bol schválený etickou komisiou Universidad Nacional de Colombia.
Izolácia a genotypizácia H. pylori
použitím štandardných metód kultivácie, ako bolo opísané skôr [17]. H. pylori
DNA bola extrahovaná z konfluentní doskových kultúr expandovaných z jedinej kolónie s použitím komerčne dostupnej súpravy (QIAGEN, Valencia, CA). Stav ČAGA
bola stanovená pomocou PCR (PCR) pre konzervované oblasti ČAGA stroje a pre priame sekvenovanie za použitia cagTF primárny pár; 5'-ACC CTA GTC GGT AAT GGG-3 'a cagTR; 5'-GCT GCT TCT TTA GAY ACY GC-3 '(Y = C alebo T) sú vyrobené 3' opakované oblasti z ČAGA
, ako bolo opísané skôr [18]. Podmienky PCR boli počiatočné denaturácia po dobu 5 min pri 95 ° C, 35 amplifikácie kroky (95 ° C počas 30 sekúnd, 56 ° C počas 30 sekúnd, a 72 ° C počas 30 s), a konečná extenzia cyklus 7 minút pri 72 ° C, za použitia Blend Taq DNA polymerázy (Toyobo, Osaka, Japan). Absencia ČAGA
bola potvrdená prítomnosť ČAGA
prázdne miesto, ako bolo opísané skôr [19]. PCR produkty boli čistené pomocou QIAquick Purification Kit (Qiagen) podľa inštrukcií výrobcu. Amplifikovanej fragment bol detekovaný pomocou elektroforézy v 1,5% agarózovom gélu, ktorý sa následne, farbené etidiumbromidom a vizualizujú pomocou ultrafialové trans-svetlo.
genotypu (S1, S2, m1 a m2) bola vykonaná ako bolo opísané skôr [20], [21]. Primery pre región signálu sa získa fragment o 259 bp a 286 bp pre S1 a S2 varianty, resp. Primery pre stredné oblasti poskytla fragment o veľkosti 570 bp a 645 bp pre M1 a M2 variantov, resp.
fylogeneticky analýza H. Pylori
kmene
) bola stanovená pomocou PCR na báze sekvenovanie, ako bolo opísané skôr [7 ], [22]. Priame sekvenovania DNA sa vykonáva za použitia AB 3130 Genetic Analyzer (Applied Biosystems, Foster City, CA) podľa inštrukcií výrobcu. Pre konštrukciu fylogenetického stromu na základe MLST genotypov, sekvenčné súbory údajov 7 domácnosti gény hpAfrica2, hpAfrica1, hpNEAfrica, hpEurope, hpSahul, hpAsia2, hspMaori, hspAmerind a hspEAsia kmeňov (60, 181, 61, 566, 49, 17, , 80, 18, a 177 kmeňov, v danom poradí, 1,209 kmene celkom) boli získané z databázy PubMLST (http://pubmlst.org/). Tieto sekvencie dátové súbory boli spojené s našimi dátami z 66 kolumbijských kmeňov. Susedné spojovacie stromy boli postavené MEGA 5,0 pomocou Kimura-2 parametre [23].
Analýza štruktúry obyvateľstva v H. pylori
kmene
-pozitívne a 11 ČAGA
-negativní. Dvadsať deväť boli Vaca
s1m1 genotyp a 6 boli Vaca
s2m2 genotyp. Všetky 24 ČAGA
-pozitívnych kmene GC ukázala, že Vaca
s1m1 genotyp. U 31 kmeňov z pacientov DU, 22 kmene boli ČAGA
-pozitívne a zvyšných deväť bolo ČAGA
-negativní. Vaca
s1m1 genotyp bol zistený u 16 kmeňov, Vaca
s1m2 5 kmeňov a Vaca
s2m2 v 10 kmeňov. Medzi 22. ČAGA
-pozitívnych kmene od pacientov DU, 14 kmeňov vlastnil Vaca
s1m1 genotyp. Päť kmene boli s1m2 a zvyšné 3 kmene boli s2m2.
stav kmeňov na strome MLST. Kmene z ČAGA
-pozitívne a ČAGA
-negativní nemožno rozdeliť jasne, hoci ČAGA
negatívnych kmene boli relatívne združené v rovnakom odbore. Obrázok 2B ukazuje Vaca
typy kmeňov na rovnakom MLST stromu. Šestnásť Vaca
s2m2 kmene boli zoskupené v sub-pobočky spolu s dvoma s1m2 a šiestimi s1m1 kmeňov. Zvyšné tri Vaca
s1m2 kmene boli umiestnené medzi 39 Vaca
s1m1 kmeňov.
-pozitívne kmeň bol umiestnený medzi pododbory hpAfrica1. Zostávajúcich 65 kmene boli rozptýlené medzi hpEurope pododborov bez ohľadu na typ ochorenia. Preto žiadny vzťah nebol pozorovaný medzi odbočenie fylogenetického stromu a klinickými výsledkami.
typy a choroby sú zobrazené značkami pod stĺpcového grafu. Ako Tento obrázok ukazuje, ČAGA
negatívnych kmene boli často na pravom konci. Zatiaľ čo väčšina z kolumbijskej kmeňov mal určité komponenty a /alebo bežnej populácii komponent hpEurope, jeden kmeň vykazovali vyššie podobnosť s hpAfrica1 (Obrázok 4C, baru označený trojuholníkom). Tento kmeň zodpovedá tej, ktorá patrí do hpAfrica1 sub-pobočky v fylogenetického stromu MLST (obrázok 3).
Diskusia
sú spojené s rôznou výsledky H. pylori
infekciu. Všeobecne platí, že rozdelenie výskytu GC úzko súvisí sa tieto H. pylori
skupiny definované na základe analýzy populačnej na základe MLST [25]. Vysoký výskyt GC bola zistená v oblastiach, v ktorých kmene hpEastAsia prevládajú (najmä hspEAsia). Avšak, výskyt GC je v Afrike, kde väčšina kmeňov sú hpNEAfrica, hpAfrica1 alebo hpAfrica2, a v južnej Ázii, kde väčšina kmeňov sú hpAsia2 veľmi nízka. Celkovo nízky výskyt rakoviny žalúdka v africkej a juhoázijské krajiny by mohla byť vysvetlená, aspoň čiastočne, podľa rôznych genotypov H. pylori
cirkulujúci v rôznych geografických oblastiach. Je zaujímavé, že nedávna správa o ČAGA
-pozitívnych kmene v Kolumbii ukázala, že všetky kmene z vysoko rizikových oblastí GC patrí k hpEurope, zatiaľ čo hpEurope a hpAfrica1 kmene koexistovať v oblastiach s nízkym rizikom [15]. Navyše, predmety infikované hpEurope kmene H. pylori
vykazovali vyššie histopatologické skóre ako nakazených kmene hpAfrica1. Autori dospeli k záveru, že rozdiel v bakteriálnej populácie môže byť použitý ako prediktor rizika GC.
kmene izolované od kolumbijskej pacientov s GC a DU ale nie gastritídu. U pacientov s gastritídou iba v dobe endoskopia sa môže vyvinúť DU a GC v neskoršom veku; Naopak, u pacientov DU zriedka sa GC počas svojho života [4], [26]. Preto, aj keď H. pylori
infekcie podporuje rozvoj ako GC a DU, GC a DU sú považované za výsledky varianty. V tejto štúdii sme nezistili žiadny rozdiel v fylogenetických skupín medzi GC a kmene DU. Bolo zistené, naše kolumbijskej kmene, že patrí do hpEurope, s výnimkou jedného kmeňa hpAfrica1. Aj keď kolumbijskej kmene boli rozdelené do niekoľkých čiastkových vetiev v fylogenetických stromov, neexistuje žiadny vzájomný vzťah medzi vetvami a chorôb (obrázky 1 a 3). Z tohto dôvodu, topológia stromu MLST nepôsobí ako marker pre vyhodnotenie GC a riziko pre DU hpEurope kmeňov v Kolumbii. V skutočnosti, dokonca aj v štúdii vykonávané de Sablet et al., To všetko ČAGA
negatívnych kmene boli tiež považujú za patriace do hpEurope [15]. Autori uviedol, že predmety infikované ČAGA
negatívnych kmeňov malo veľmi nízke histopatologické skóre; Preto, hpEurope kmene bez prítomnosti ČAGA
môže byť menej virulentné. Okrem toho sme už skôr oznámil, že kmene s viac ako tromi opakovaných oblastí regiónu 3 'v ČAGA
boli spojené s výrazne vyšším skóre pre žalúdočné slizničnej atrofie a črevné metaplázia, než tie s menším počtom opakovaných regióny v Kolumbii [20 ]. Prevalencia kmeňov s viac ako tromi repetície bola vyššia ako GC DU [20]. To znamená, že typ ČAGA
skôr než phylogeographic pôvodu, môže byť lepšie predpovedať GC pre kmeňov hpEurope [27].
PAI sú spojené iba OipA bol nezávislým rizikovým faktorom pre DU [28]. OipA v ČAGA
-pozitívnych kmene môžu prispieť k phylogeographic variante určenej MLST analýzy. Okrem toho, hlásil sme, že jhp0045 stroje a jhp0046
boli významne spojené s GC v ČAGA
-pozitívnych kmene v Kolumbii [29]. Okrem toho, hlásil sme, že infekcia s Dupa
-pozitívnych kmene zvyšuje riziko pre Du ale boli jej účinnosť proti GC v Kolumbii [30]. Z tohto dôvodu, je stav iné faktory virulencie, môže byť v korelácii s fylogenetického stromu získa podľa MLST [5]. Vzťahy medzi fylogenetický domácnosti génov a CAG
PAI fylogenetický boli hlásené [31]. Phylogeny väčšiny CAG
PAI gény bol podobný tomu z MLST, čo ukazuje, že CAG
PAI bol pravdepodobne získal iba raz H. pylori stroje a jeho genetická diverzita odráža izoláciu podľa vzdialenosti, ktorá má tvar tento bakteriálnych druhov, pretože moderné ľudia sťahovali z Afriky [31]. V tejto štúdii sa fylogenetický strom založený na MLST nie je v korelácii s typom ČAGA
alebo krava
, aj keď kmeňov s ČAGA
-negativní alebo krava
s2m2 genotypy boli relatívne klastrov. Iba jeden kmeň patril hpAfrica1, a tento kmeň vlastnil ČAGA
, čo naznačuje, že ČAGA stroje a Vaca
genotypy neboli určujúce pre fylogenetického pôvodu z MLST v andskom región v Kolumbii. Avšak, sme už skôr oznámil, že typ ČAGA
bola spojená s fylogeneticky klastra v Okinawa, Japonsko [22], [27]. Kmene z Okinawy boli rozdelené do 3 podskupín a raz prímesou skupiny ovplyvnené západnými kmene. Phylogeographic topológie a subpopulácie boli významne spojené s klinickými výsledkami; Avšak rozdiel v phylogeographic topológii bol kvôli stavu ČAGA
(typ East-Asian, západného typu ČAGA stroje a ČAGA
-negativní) a Vaca
(m1 alebo m2) z čiastkových populácií. GC bol viac prevláda v klastri, ktorý obsahuje prevažne východoázijských-type ČAGA
kmeňov. Výsledky MLST ukazujú, že prevalencia GC v každej skupine nelíšili, keď jedinou západnej typ ČAGA
kmene alebo ČAGA
negatívnych kmeňov boli použité [27]. Tak phylogeographic pôvode môžu pôsobiť ako za pozoruhodný faktor v predpovedanie faktory virulencie, ale nie výsledok ochorenia (napr, v hspEAsia, väčšina z nich je ČAGA
-pozitívny, Vaca
S1 /M1 typ baba
-pozitívne a oipA
"on" stavu). V nedávnej štúdii, a to nielen H. pylori
pôvod, ale aj koevoluce medzi človekom a H. pylori
bolo zistené, že najlepším ukazovateľom pre prekanceróznych lézií [32]. Ďalšie štúdie sú nutné potvrdiť vzťah medzi rodičovstva H. pylori
bez ohľadu faktorov virulencie a klinické výsledky.
izolovaný od mesticov by zobraziť zmiešaný typ hpEurope a hspAmerind. Je zaujímavé, že vo všetkých prípadoch okrem jedného v našej štúdii boli infikované hpEurope kmeňov, v súlade s predchádzajúcou štúdiu [15]. štruktúrne analýza odhalila, že niektoré kmene s hpEurope ukázal mozaika vzorku; Avšak, oni neboli zmes hpEurope a hspAmerind. Toto zistenie podporuje hypotézu, že kmene hpEurope vlastniť konkurenčnú výhodu oproti domorodých kmeňov hspAmerind, ako bolo opísané skôr [34] [35]. Prekvapivo, naše predchádzajúce štúdia ukázala, že aj v 16 kmeňov izolovaných z Huitoto, primitívne, izolované skupiny žijúci v amazonských džungliach v Kolumbii, 4 kmene boli infikované hspAmerind, zatiaľ čo zvyšných 12 bolo hpEurope kmeňov [12]. Navyše sme zistili, že niektoré indiánske kmene z Kolumbie vlastnil západného typu ČAGA
ale nie východoázijských-type ČAGA
[36]. Zostáva nejasné, prečo väčšina Amerindian H. pylori
kmene majú genotypy typické kmeňov zo západných krajín, a to aj v miestach, kde sa zdá byť minimálny západnej vplyv. hpEurope, skôr než kmene hspAmerind, mohlo by ľahko prispôsobiť podmienkam v oblasti životného prostredia a selektívne tlak vyvíjaný hostiteľom. Dve hypotézy, jeden vychádzajúci z kmeňa súťaže a ďalšie na kmeň podvracanie transformáciou, boli navrhnuté ako dôvody pre posunutie hspAmerind podľa hpEurope [34]. Ďalšie štúdie bude potrebné definovať mechanizmus (y) zodpovedný za in vivo
strata Amerindians kmeňov v konkurencii s kmeňmi Starého sveta. Zaujímavé je, že sme zistili, že niektoré kolumbijskej kmene vykazovali určitú zložku obyvateľstva, a táto zložka bola zdieľaná nielen s ostatnými juhoamerickými kmeňov, ale tiež so španielskymi kmeňov uložených v PubMLST. Mesto Bogota, ktorá bola založená v roku 1538, sa stal jedným z hlavných administratívnych centier španielskej dŕžavy v Novom svete (spolu s Lima a Mexico City) [33]. Naša analýza naznačuje, že hpEurope kmene v Kolumbii boli zavedené hlavne španielskych prisťahovalcoch. Populácia typizácie H. pylori
je užitočným nástrojom pre vzory mapovanie ľudskej migrácie [37]; Naozaj, naše zistenie by mohlo byť užitočné pre objasnenie tých moderných ľudí v Južnej Amerike. Hoci naše analýza MLST na základe siedmich domácnosti génov naznačuje, že väčšina z kolumbijskej kmene boli nahradené kmene hpEurope, zvyšky rodových pôvodných kmeňov by mohli zostať v iných častiach genómu. Rozsiahlejšie po celom svete a celého genómu prieskumy nám pomôže ďalej porozumieť vývoju a populačný štruktúru H. pylori
.
boli izolované. Vskutku, bolo navrhnuté, že hostiteľ pôvod môže mať vplyv na klinické výsledky, ako je popísané vyššie [32]. Napríklad, zápalových génov cytokínov polymorfizmy ( IL-1
zoskupenie génov, TNF-α
IL-10 stroje a IL-8
) boli hlásené byť v korelácii s GC [38] - [43]. Okrem toho, v rodinnej anamnéze GC bolo preukázané, že prispieva ku klinickým výsledkom [44]. Okrem iného H. Pylori
virulentné faktory, môžu byť užitočné pre rozlišovanie medzi GC a rizikom DU. Ďalšie vyšetrovanie je potrebné objasniť vzťah medzi H. pylori
genotypy a výsledky choroby.
môže byť potrebné presnejšie určiť klinické výsledky. Navyše, naša analýza tiež naznačuje, že hpEurope kmene v Kolumbii boli zavedené predovšetkým španielskych prisťahovalcoch a ich genotyp nebol ovplyvnený Amerindians.
Podporné informácie
tabuľke S1.
doi: 10,1371 /journal.pone.0105392.s001
(DOCX)
.
 Molekuly mikróbov proti koronavírusu by mohli byť kľúčom k novej liečbe
Molekuly mikróbov proti koronavírusu by mohli byť kľúčom k novej liečbe
 Prečo by ste mali do stravy zaradiť prírodné zdroje vlákniny
Prečo by ste mali do stravy zaradiť prírodné zdroje vlákniny
 Štúdia objasňuje príčiny oslabujúcej bolesti čriev
Štúdia objasňuje príčiny oslabujúcej bolesti čriev
 Mikrobióm spermií odhalený sekvenovaním RNA
Mikrobióm spermií odhalený sekvenovaním RNA
 Nanotechnológia a diagnostika a liečba COVID-19
Nanotechnológia a diagnostika a liečba COVID-19
 Rýchle občerstvenie môže byť hlavným vinníkom depresie mladistvých
Rýchle občerstvenie môže byť hlavným vinníkom depresie mladistvých
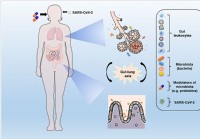 Mikrobiotická modulácia a obnovenie eubiózy by mohli pomôcť obmedziť komplikácie súvisiace s COVID-19
Koronavírusová choroba (COVID-19), spôsobená závažným akútnym respiračným syndrómom koronavírusom 2 (SARS-CoV-2), postihuje mnoho orgánov v tele. Okrem toho, že ide o ochorenie dýchacích ciest, môže t
Mikrobiotická modulácia a obnovenie eubiózy by mohli pomôcť obmedziť komplikácie súvisiace s COVID-19
Koronavírusová choroba (COVID-19), spôsobená závažným akútnym respiračným syndrómom koronavírusom 2 (SARS-CoV-2), postihuje mnoho orgánov v tele. Okrem toho, že ide o ochorenie dýchacích ciest, môže t
 Krvný test na mikrobiálnu DNA môže varovať pred rakovinou
Nová štúdia publikovaná v časopise Príroda 11. marca 2020, by mohlo zmeniť v súčasnosti zastávané názory na príčinnú súvislosť a diagnostiku rakoviny. Vedci prišli s novou technikou na zisťovanie pr
Krvný test na mikrobiálnu DNA môže varovať pred rakovinou
Nová štúdia publikovaná v časopise Príroda 11. marca 2020, by mohlo zmeniť v súčasnosti zastávané názory na príčinnú súvislosť a diagnostiku rakoviny. Vedci prišli s novou technikou na zisťovanie pr
 Závažné komplikácie COVID-19 súvisiace s poruchou črevnej bariéry
Hlavným zameraním výskumu súčasnej pandémie koronavírusovej choroby 2019 (COVID-19) bola potreba porozumieť fungujúcim mechanizmom, ktoré spôsobujú rôzne prejavy a komplikácie tejto potenciálne smrteľ
Závažné komplikácie COVID-19 súvisiace s poruchou črevnej bariéry
Hlavným zameraním výskumu súčasnej pandémie koronavírusovej choroby 2019 (COVID-19) bola potreba porozumieť fungujúcim mechanizmom, ktoré spôsobujú rôzne prejavy a komplikácie tejto potenciálne smrteľ