Um die Muster der Genexpression bei Magenkrebs untersuchen, insgesamt 26 gepaart Magenkrebs und noncancerous Gewebe von Patienten für die Genexpression Microarray-Analysen eingeschrieben waren. Limma Methoden wurden angewandt, um die Daten zu analysieren, und Gene wurden als signifikant differentiell exprimiert werden, wenn der False Discovery (FDR) Wert war < 0,01, P Citation:. Li H Yu B, Li J, Su L, Yan M, Zhang J, et al. (2015) Charakterisierung von differentiell exprimierten Gene beteiligt in Pathways Verbunden mit Magenkrebs. PLoS ONE 10 (4): e0125013. doi: 10.1371 /journal.pone.0125013 Academic Editor: Francisco J. Esteban, Universität Jaén, Spanien Empfangen: 9. November 2014; Akzeptiert: 6. März 2015; Veröffentlicht: 30. April 2015 Copyright: © 2015 Li et al. Dies ist ein offener Zugang Artikel unter den Bedingungen der Lizenz Creative Commons Attribution verteilt, die uneingeschränkte Nutzung erlaubt, die Verteilung und Vervielfältigung in jedem Medium, vorgesehen sind der ursprüngliche Autor und Quelle genannt Datenverfügbarkeit: Alle relevanten Daten sind innerhalb des Papiers und seiner Hintergrundinformationen Dateien. Alle Microarray-Dateien sind vom NCBI Gene Expression Omnibus (GEO) Datenbank (Zugangsnummer "GSE65801"; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE65801) zur Verfügung. die Finanzierung: Diese Arbeit wurde durch Zuschüsse für die Analyse von der National Natural Science Foundation of China unterstützt wurde [Nr 81172324, Nr 91229106, Nr 81272749 und Nr 81372231], Wissenschaft und Technologie Kommission von Shanghai Gemeinde [Nr 13ZR1425600] und Schlüsselprojekte in der National Science & Technologie Säulen-Programm von China (Nr 2014BAI09B03). Die Geldgeber hatten keine Rolle in Studiendesign, Datenerfassung und Analyse, Entscheidung oder Vorbereitung des Manuskripts zur Veröffentlichung Konkurrierende Interessen:.. Die Autoren haben erklärt, dass keine Interessenkonflikte bestehen Einführung Magenkrebs (GC) ist eine der häufigsten Krebserkrankungen weltweit, und die Inzidenz ist besonders hoch in Ost-Asien, insbesondere in China. Etwa 952.000 neue Fälle von Magenkrebs wurden weltweit im Jahr 2012 diagnostiziert, und die Hälfte von ihnen traten in Ostasien (vor allem in China) [1]. In China werden die Mehrzahl der Patienten mit GC in einem späten Stadium mit schlechter Prognose festgestellt. Daher Aufklärung der molekularen Mechanismen GC Progression ist wichtig, um die Ermittlung der wichtigsten Biomarker und die Entwicklung wirksamer zielgerichtete Therapien zugrunde liegen. Im Laufe der letzten zehn Jahre, die Genexpression Microarrays ein gemeinsames Werkzeug für die Untersuchung Gentranskriptes Ebenen in der Krebsforschung geworden sind. Mikroarray-Daten für eine Vielzahl von Analysen, wie beispielsweise unbeaufsichtigten clustering, Klassifizierung, differentielle Expressionsanalyse und die Expression Mapping quantitative trait loci verwendet [2]. Es hilft nicht nur Schlüssel dysfunktionalen Gene in der Krebs zu identifizieren, sondern bietet genomweite Informationen über die Genexpression zu einer Zeit als auch [3,4]. In dieser Studie haben wir eine genomweite Umfrage der Expression von lncRNAs und mRNAs aus gepaarte Stichproben von primären Magenkrebsgewebe und noncancerous Gewebe, die unterschiedlich exprimiert lncRNAs und kodierenden RNAs zu profilieren. Untersuchung dieser Daten werden wertvolle Informationen über den Mechanismus der Karzinogenese bieten und Entdeckung von Schlüsselgenen erlauben, die als Ziele für die Zukunft der Anti-Krebs-Therapie wirken. Ethical Aussage Eine schriftliche Einverständniserklärung wurde von allen Teilnehmern erhalten. Die Studie wurde von der Human Research Ethics Committee von Ruijin Hospital, Shanghai Jiao Tong University, School of Medicine zugelassen. Die Gewebe wurden aus primären Magenkarzinomen von unbehandelten Patienten genommen, die D2 unterzog radikale Gastrektomie in Shanghai Ruijin Hospital. Für jeden Krebsgewebe wurde eine gepaarte noncancerous Gewebeprobe aus dem benachbarten Bereich zur gleichen Zeit gesammelt. Die Größe jeder Probe war etwa 0,1 cm 3. Alle Proben wurden in RNAlater innerhalb von 15 Minuten nach der Exzision und in flüssigem Stickstoff gelagert, bis RNA-Extraktion platziert. In dieser Studie wurden 32 gepaarte Gewebe für die Microarray gesammelt und 26 gepaarte Stichproben wurden für die nächste Schritt Analyse von GO, Bahn und Netz nach Qualitätskontrolle mit Hilfe von 3D Hauptkomponentenanalyse (3D-PCA) und Cluster-Analyse aufgenommen. Microarray-Experimenten Agilent Sureprint G3 Menschliche GE 8x60K Microarray (Design-ID: 028004) wurde in dieser Studie eingesetzt. Die Gesamt-RNA wurde isoliert und verstärkt, um ein Low Input Schnell Amp Labeling Kit, One-Color (Catȇ0-2305, Agilent Technologies, US). Dann wurden die markierten cRNAs durch eine RNeasy Mini Kit (Cat˥06, QIAGEN, Deutschland) gereinigt. auf die Anweisungen des Herstellers Basierend wurde jede Folie mit 600 ng Cy3-markierten cRNA hybridisiert ein Gene Expression Hybridization Kit (CatȆ8-5242, Agilent Technologies, USA) und gewaschen durch die Gene Expression Wash Buffer Kit (CatȆ8-5327, Agilent Technologies, US). Ein Agilent Microarray-Scanner (Cat # G2565CA, Agilent Technologien, US) und Feature Extraction Software 10.7 (Agilent Technologies, wurden US) angewendet jede Folie mit den gleichen Einstellungen wie folgt, Dye-Kanal gezeigt scannen: Grün, Scanauflösung = 3 um, 20bit. Die Rohdaten wurden von der Quantile Algorithmus, Gene Frühling Software 11.0 (Agilent Technologies, US) (detailliert in S5 Tabelle). Lineare Modelle und empirische Bayes Methoden normalisiert wurden angewandt zu analysieren die Daten in dieser Studie. Die resultierenden P GO Kategorie ausgeführt Wir Gene Ontology (GO) analysiert die Funktionen der differentiell exprimierten Gene in unserem Microarray-Analyse nach dem Schlüssel funktionelle Klassifizierung des National Center for Biotechnology Information (NCBI). Im Allgemeinen exakten Fisher-Test und der χ Pathway-Analysen Pathway Anmerkungen der Differential exressed Gene wurden aus KEGG (http erhalten: //www .genome.jp /kegg /). Pathway Kategorien mit einem FDR < 0,01 markiert wurden. Die Anreicherung von signifikanten Wege wurde gegeben durch: Anreicherung = /, die uns geholfen, mehr signifikante Wege in unserer Studie zu lokalisieren ( n Gene-Act Netzwerk nach der KEGG-Datenbank, ein Gen in mehreren Bahnen beteiligt sein können oder mit mehreren anderen Genen interagieren. Alle Gen-Gen-Interaktionen wurden gepoolt das Gene-Act Netzwerk aufzubauen auf der Grundlage der Differenzwege, die uns geholfen, die Signalwege und wichtige regulatorische Gene in GC offenbaren. Gene Coexpression Netz wurde entsprechend dem normalisierten Signalintensität von spezifischen Expressionsgenen aufgebaut. Grad Zentralität ist als die Anzahl der Verbindungen definiert, einem Knoten zum anderen hat, was die relative Bedeutung von Genen bestimmt. Was mehr ist, k-Kerne wurden als Verfahren zur Vereinfachung der Graph Topologie Analysen angewandt. Kern regulatorische Faktoren (Gene), die haben die höchsten Grade verbinden die meisten benachbarten Genen und bauen die Struktur des Netzwerks (die in S5 Tabelle). Die Gesamt-RNA extrahiert wurde aus Gewebe des Trizol-Reagens (Invitrogen) gemäß den Anweisungen des Herstellers verwendet wird. Die quantitative Echtzeit-Polymerase-Kettenreaktion (PCR) wurde in einem Fast Real-Time-PCR-System 7500 (Applied Biosystems) unter Verwendung von SYBR-Grün PCR Master Mix durchgeführt. Die Primer der 10 Gene wurden in S4 Tabelle gezeigt. PCR-Reaktionen wurden bei 50 ° C für 2 min durchgeführt, gefolgt von 40 Zyklen von 95 ° C für 15 s und 60 ° C für 1 min. ACt wurde durch Subtrahieren des von Ct β-Actin-RNA (Kontrolle) aus dem Ct der RNA der Probe, jeweils berechnet. ΔΔCt wurde dann durch Subtrahieren des ACt der Steuerung von der ACt der Probe berechnet. Falten Sie Änderung durch die Gleichung 2-ΔΔCt berechnet wurde. SPSS-Software 19 und Microsoft Excel 2010 verwendet wurde, um die Daten zu analysieren. Die Expressionsniveaus zwischen Krebsgewebe und angrenzenden noncancerous Gewebe wurden durch gepaarte Stichproben-t-Tests analysiert. P
-Wertes WAS < 0,01 und die Fold-Change (FC) war > 2. Anschließend wurden Gene Ontology (GO) entsprechend den Hauptfunktionen der differentiell exprimierten Gene zu analysieren. Nach dem Kyoto-Enzyklopädie von Genen und Genomen (KEGG) Datenbank fanden wir Wege signifikant mit den differentiellen Genen assoziiert. Gene-Act-Netzwerk und Coexpression Netzwerk aufgebaut wurden jeweils auf der Grundlage der Beziehungen zwischen den Genen, Proteinen und Verbindungen in der Datenbank. 2371 mRNAs und 350 lncRNAs als signifikant differentiell exprimierten Gene betrachtet wurden für die weitere Analyse ausgewählt. Die Kategorien GO, Weg analysiert und das Gen-Act Netzwerk zeigten ein konsistentes Ergebnis, dass hochregulierte Gene, die für die Tumorentstehung, Migration, Angiogenese und Mikro Bildung, während nach unten reguliert Gene verantwortlich waren im Stoffwechsel beteiligt waren. Diese Ergebnisse dieser Studie bieten einige neue Erkenntnisse über kodierende RNAs, lncRNAs, Wege und die Co-Expression Netzwerk bei Magenkrebs, die nützlich sein wird, weitere Untersuchungen zu führen und Ziel Therapie für diese Krankheit
Methoden und Materialien
Gewebeproben
Limma
-Werten wurden unter Verwendung des BH FDR-Algorithmus angepasst. Es gab drei Standards für uns, dass ein Gen zu betrachten war signifikant unterschiedlich exprimiert, die FDR-Wert betrug < 0,01, P
-Wertes WAS < 0,01 und die Falte Änderung war > 2. (Detailliert in S5 Tabelle)
2 Test angewandt wurden die GO Kategorie zu klassifizieren, und die falsche Discovery Rate (FDR,) berechnet die P
zu korrigieren -Wertes ( N
k
bezieht sich auf die Anzahl der Fisher-Test P
-Werten kleiner als die χ
2 Test P
-Werten). Die Anreicherung Re wurde gegeben durch: Re = ( n
f
/ n
) /( N
f
/ N
) in den wesentlichen Kategorien ( N
f Was ist die Anzahl der Differential Gene innerhalb der jeweiligen Kategorie, n Was ist die Gesamtzahl der Gene innerhalb der gleichen Kategorie, n
f Was ist die Anzahl der Differential Gene im gesamten Mikroarray, und N
ist die Gesamtzahl der Gene in dem Microarray) (in S5 Tabelle beschrieben)
g Was ist die Anzahl der Differential Gene innerhalb des insbesondere Weg, n
a Was ist die Gesamtzahl der Gene innerhalb des gleichen Weg, N
g Was ist der Anzahl von Differenz Genen, die mindestens einen Stoffwechselweg Annotation haben und N
a was ist die Anzahl von Genen, die mindestens einen Stoffwechselweg Annotation in der gesamten Mikroarray aufweisen.) (detailliert in S5 Tabelle).
Die Co-Expression Netzwerk
Real-time quantitative PCR
Die statistische Analyse
-Werten unter 0,05 als statistisch signifikant angesehen wurden.
Ergebnisse
 Mathematisches Modell zeigt das Risiko einer SARS-CoV-2-Infektion nach fäkaler Mikrobiota-Transplantation
Mathematisches Modell zeigt das Risiko einer SARS-CoV-2-Infektion nach fäkaler Mikrobiota-Transplantation
 COVID-19:Wie die Krankheit aussieht
COVID-19:Wie die Krankheit aussieht
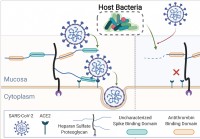 Menschliches Mikrobiom trimmt Schleimhautglykane,
Menschliches Mikrobiom trimmt Schleimhautglykane,
 Neuartige Wirt-Virus-Mikrobiom-Interaktionen während COVID-19 können das Ergebnis bestimmen
Neuartige Wirt-Virus-Mikrobiom-Interaktionen während COVID-19 können das Ergebnis bestimmen
 Fortfahren,
Fortfahren,
 Wissenschaftler lösen mysteriösen Fall von Auto-Brauerei-Syndrom
Wissenschaftler lösen mysteriösen Fall von Auto-Brauerei-Syndrom
 Elektronische Pille zur Gaserkennung zur Diagnose von Magen-Darm-Beschwerden
Wissenschaftler der RMIT Universität, Melbourne, haben eine elektronische Pille entwickelt, die spezielle Gase im Darm erkennen und Ärzten bei der Diagnose von Magen-Darm-Beschwerden wie dem Reizdarms
Elektronische Pille zur Gaserkennung zur Diagnose von Magen-Darm-Beschwerden
Wissenschaftler der RMIT Universität, Melbourne, haben eine elektronische Pille entwickelt, die spezielle Gase im Darm erkennen und Ärzten bei der Diagnose von Magen-Darm-Beschwerden wie dem Reizdarms
 Gute Nachrichten für RDS-Betroffene, da Forscher „Darmjucken“ feststellen
Forscher der Flinders University am South Australian Health and Medical Research Institute haben eine wichtige Entdeckung über die Schmerzen gemacht, die bei Reizdarmsyndrom (IBS) auftreten.
Gute Nachrichten für RDS-Betroffene, da Forscher „Darmjucken“ feststellen
Forscher der Flinders University am South Australian Health and Medical Research Institute haben eine wichtige Entdeckung über die Schmerzen gemacht, die bei Reizdarmsyndrom (IBS) auftreten.
 Forscher identifizieren in vitro ein Bakterium mit Anti-SARS-CoV-2-Aktivität:Dolosigranulum pigrum
Das schwere akute respiratorische Syndrom Coronavirus 2 (SARS-CoV-2) hat weltweit über 173,3 Millionen Menschen infiziert. Von diesen, eine bedeutende Minderheit war schwerwiegend oder kritisch, weltw
Forscher identifizieren in vitro ein Bakterium mit Anti-SARS-CoV-2-Aktivität:Dolosigranulum pigrum
Das schwere akute respiratorische Syndrom Coronavirus 2 (SARS-CoV-2) hat weltweit über 173,3 Millionen Menschen infiziert. Von diesen, eine bedeutende Minderheit war schwerwiegend oder kritisch, weltw