Aberrant miRNA-Expression moduliert abnorm Genexpression in den Zellen und kann zu tumorigenesis beim Menschen beitragen. Diese Studie funktionell relevante differentiell exprimierten Gene mit den Transkriptionsfaktoren und miRNA-co-regulierte Netzwerkanalyse für Magenkrebs identifiziert. Der TF-miRNA koregulative Netzwerk wurde aus cDNA-Microarray und miRNA-Expressionsprofilerstellung von Magenkrebsgewebe erhalten auf Basis von Daten aufgebaut. Das Netzwerk zusammen mit ihren Co-regulierte Gene wurde mit Datenbank für Annotation, Visualisierung und integrierten Entdeckung (DAVID) und Transkriptions-regulatorische Element-Datenbank (TRED) analysiert. Wir fanden achtzehn (17 hochreguliert und 1 herunterreguliert) differentiell exprimierten Genen, die von Transkriptionsfaktoren und miRNAs co-reguliert waren. KEGG Pathway-Analyse ergab, dass diese Gene ein Teil der extrazellulären Matrix-Rezeptor-Wechselwirkung und focal adhesion Signalwege waren. Darüber hinaus zeigte qRT- PCR und Western-Blot-Daten einen Anstieg in COL1A1 und Abnahme in NCAM1 mRNA und Proteinspiegel in Magenkrebsgewebe. Somit können, sofern diese Daten die ersten Beweise zu zeigen, dass veränderte Gen-Netzwerk mit Magenkrebs Invasion assoziiert war. Weitere Studien mit einer großen Probengröße und funktioneller Experimente ist notwendig, um diese Daten zu bestätigen und zu Diagnose- und Behandlungsstrategien für Magenkrebs beitragen
Citation:. Shi Y, Wang J, Xin Z, Duan Z, Wang G , Li F (2015) Transkriptionsfaktoren und microRNA-Co-regulierte Gene in Magenkrebs Invasion in Ex Vivo Academic Editor: Jian-Jun Zhao, Dana-Farber Cancer Institute, UNITED STATES Empfangen: 8. November 2014; Akzeptiert: 24. Februar 2015; Veröffentlicht: 10. April 2015 Copyright: © 2015 Shi et al. Dies ist ein offener Zugang Artikel unter den Bedingungen der Lizenz Creative Commons Attribution verteilt, die uneingeschränkte Nutzung erlaubt, die Verteilung und Vervielfältigung in jedem Medium, vorgesehen sind der ursprüngliche Autor und Quelle genannt Datenverfügbarkeit: Alle relevanten Daten sind innerhalb des Papiers und seiner Hintergrundinformationen Dateien Finanzierung:. Diese Arbeit wurde unterstützt durch Zuschüsse von der National Natural Science Foundation of China (̭20108025 und̮72662) unterstützt. Es wird auch teilweise durch National Natural Science Foundation of China (̬71897 und̮01712), Jilin Key Laboratory of Biomedical Materials, Gründung der Provinz Jilin Abteilung Wissenschaft und Technologie (É30522013JH undÉ40414048GH) und dem Norman Bethune Programm von Jilin unterstützt Universität (É2219) konkurrierende Interessen:.. die Autoren haben erklärt, dass keine Interessenkonflikte bestehen Einführung Magenkrebs ist eine der häufigsten Form von malignen Erkrankungen in die Welt, die zu einem Drittel der krebsbedingten Todesfälle bei Männern beitragen und ein Fünftel der Frauen [1]. Etwa treten zwei Drittel der Magenkrebsfälle in den Entwicklungsländern. In China ist die Inzidenz und Mortalität an Magenkrebs im Zusammenhang mit den dritten Platz unter den anderen Formen von bösartigen Erkrankungen [2] und es wurde berichtet, dass Magenkrebs häufiger in ländlichen Gebieten und mit einem Trend der jüngeren Menschen beeinflusst zu werden, indem sie es in den letzten Jahren tritt [3 ]. Umwelt (wie Helicobacter pylori Die Genexpression in Zellen sowohl bei der Transkription und post-Transkriptionsebenen gesteuert. Transkriptionsfaktoren (TFs) Gentranskription koordinieren, während miRNAs Gen-Expression reguliert durch posttranskriptionales Ereignisse zu vermitteln, wie mRNA-Abbau und Proteintranslation [5]. Daher kann jede Änderung in miRNA Funktion führen bei der Entwicklung von Krebs beim Menschen [6,7]. Transkriptionsfaktoren sind Proteine, die an spezifische DNA-Sequenzen binden, die Rate der Transkription der genetischen Information von der DNA zu mRNA [8,9] zu steuern, während miRNAs eine Gruppe von einer kleinen nicht-kodierenden RNA in Zellen und die Funktion in RNA silencing und Post sind -transcriptional Regulation der Genexpression [10,11]. Der TF-miRNA Genregulationsnetz bestimmt den Genexpressionsprofil in Zellen zu einem gewissen Grad. Daher Analyse der TF-miRNA Co-regulatorischer Netzwerke in Magenkrebsgewebe könnte uns helfen, unser Verständnis zu fördern, wie TFs und miRNAs die Regulation der Genexpression koordinieren, um Magen-Krebsentstehung beitragen [12]. In unserer früheren Studie, profilierte wir differentiell exprimierte Gene in achtzig Paare von Magenkarzinom benachbarten normalen Geweben unter Verwendung von cDNA-Mikroarrays [13] und eine Reihe von Genen, die mit einer veränderten Expression, einschließlich TFs gefunden. Basierend auf den Informationen von Transkriptions-regulatorische Element-Datenbank (TRED) [14], die wir gebaut und verfestigt, um einen TF-Genregulationsnetz. In dieser Studie untersuchte, wir ausgedrückt differentiell miRNAs in fünf Paaren von Magenkarzinom-Normalgewebe und ein miRNA-Ziel regulatorische Netzwerk für Magenkrebs konstruiert durch die miRNA-Targeting-Gen-Datenbanken zu integrieren, einschließlich Targetscan, MIRANDA, miRDB und miRWalk [15] . Wir bauten dann die TF-miRNA koregulative Netzwerk unserer bisherigen Daten und anschließend ausgeführt GO und KEGG Weg analysiert und ausgeführt Echtzeit-PCR und Western-Blot-Analyse dieser Daten zu überprüfen. So könnte wichtige Hinweise für künftige Studien über miRNA und TFs Funktionen bei Magenkrebs bieten sowohl der Methoden und Analysen. Gewebeproben Insgesamt 25 Magenkarzinom-Patienten wurden für diese Studie von der ersten Krankenhaus der Universität Jilin, Changchun, China rekrutiert. Magenkrebsgewebe und die passenden entfernten Nicht-Krebsgewebe wurden operativ reseziert und in flüssigem Stickstoff innerhalb von 10 Minuten nach der Resektion gespeichert. Eine schriftliche Zustimmungen wurden von allen Probanden erhalten und die Daten wurden anonym ausgewertet. Die TNM und histologischen Klassifikation wurden nach Angaben der Weltgesundheitsorganisation (WHO) Kriterien. Diese Studie wurde von der Ethikkommission der Hochschule für Medizinische Grundlagenwissenschaften, Universität Jilin genehmigt wurde. Die differentiell exprimierten mRNA Daten zwischen Magenkrebs und normales Gewebe wurde von 80 Patienten durchgeführt und berichtet vorher [13]. Wir haben ≥ 2-fache Veränderung der differentiell exprimierten Gene, die für diese Studie zu profilieren. In dieser Studie unterschiedlich exprimiert miRNAs in 5 Paaren von Magenkrebs benachbarten normalen Geweben (siehe Patientendaten in S2 Tabelle) waren unter Verwendung von Affymetrix miRNA Microarray-Chips profiliert nach den Protokollen des Herstellers. Verwendung des Mirvana miRNA Isolation Kit (Ambion, Austin, TX, USA) und dann auf Gene Chip MikroRNA Array unterworfen Kurz gesagt wurde Gesamt-RNA aus Gewebeproben isoliert wurde unter Verwendung des Trizol (Invitrogen, Carlsbad, CA, USA) und miRNA isoliert und gereinigt Analyse. Die Daten wurden unter Verwendung von GenChip Scanner3000 mit GenChip Betriebssoftware (GCOS) und analysiert gescannt. Basierend auf die GenChip Menschen Exon 1.0 ST Microarray-Daten (Affymetrix, CA, USA), konstruierten wir das TF-Gen-Netzwerk von Genexpressionsprofilen und Transkriptionsregulationselement-Datenbank (TRED) zu integrieren. Regulatorische Interaktionen zwischen microRNA und deren Zielgene wurden basierend auf den Informationen gegründet von Targetscan, MIRANDA, miRDB und miRWalk Datenbank. Die TF-miRNA Co-regulatorische Netzwerke wurden durch überlappende diese beiden Abschnitte aufgebaut. Hub-Gene, die durch TFs co-reguliert und miRNAs wurden ebenfalls identifiziert. Die Netze wurden unter Verwendung von Cytoscape Software (Institut für Systembiologie, USA, http://www.cytoscape.org) konstruiert. Online-Analyse-Tools wie Datenbank für Annotation, Visualisierung und integrierten Entdeckung (DAVID) und Kyoto Encyclopedia of Genen und Genomen (KEGG) wurden angewandt, um die funktionalen Weg zu erkunden mit differentiell exprimierten Genen in Verbindung gebracht. KEGG Wege mit p <deutlich angereichert; 0,01 wurden identifiziert und weiter analysiert. Für mRNA-Ebene erfassen, verwendeten wir 5 ug Gesamt-RNA-Proben von jeder Probe mit der in cDNA umgekehrt transkribieren Erststrang-cDNA-Synthese-Kits (Takara, Dalian, China) und dann zur Expression von COL1A1 mit qPCR verstärkt und NCAM1 mRNA mit SYBR Premix Ex Taq (Takara) in Applied Biosystems 7300 Fast Real-Time PCR-System gemäß den Anweisungen des Herstellers. Die relative Expression der mRNA-Spiegel wurde normalisiert ß-Aktin-mRNA durch vergleichende Ct Methode (2 -ΔΔCt, ACt = Ct Ziel-Ct β-Actin, ΔΔCt = ACt Tumor-ACt normal). Alle Primer wurden mit Primer Premier 6 Software, Primersequenzen für die Amplifikation in der Tabelle aufgeführt wurden 1. Daten von qRT-PCR mit GraphPad Prism Version analysiert wurden 5,0, Unterschiede zwischen den Gruppen wurden statistisch durch Probe ein-tailed Student-t-Test mit p ausgewertet Wert. < 0,05 als wesentlich Proteinextraktion und Western-Blot Gewebeproben mit 1 mm 3 in der Größe waren in flüssigem Stickstoff gemahlen und homogenisiert in einem Zell-Lyse-Puffer (Beyotime , Beijing, China) bei 4 ° C für 20 min. Die Konzentration von Protein in den Proben wurde unter Verwendung eines BCA Protein Assay Kit (Bio-Rad, Hercules, CA, USA) bestimmt und die Proteine Proben durch Natriumdodecylsulfat-Polyacrylamid-Gelelektrophorese (SDS-PAGE) unter Verwendung von 10% Gel aufgetrennt wurden, und dann auf eine PVDF-Membran übertragen (0,45 &mgr; m, Bio-Rad, Hercules, CA, USA) für 2 h. 1000, ein Maus-anti-NCAM1 /CD56-Antikörper (Novus Biologicals) in einer Verdünnung von 1:: Die Membranen wurden dann mit einem Kaninchen-anti-Kollagen-I-Antikörper (Novus Biologicals, Littleton, CO, USA) in einer Verdünnung von 1 inkubiert 400 oder ein Kaninchen-anti-β-Actin-Antikörper (Proteintech, Chicago IL, USA) in einer Verdünnung von 1: 2000 bei 4 ° C über Nacht und anschließend mit Tris-basierten saline-Tween 20 (TBST) nach dem Waschen wurden die Membranen inkubiert mit einem Ziege-anti-Kaninchen-IgG (Beyotime) oder Ziege-anti-Maus-IgG (Proteintech) bei einer Verdünnung von 1: 2000 für 2 h. Die Protein-Signale wurden durch Autoradiographie nachgewiesen durch erhöhte Chemilumineszenz-Reagenz (Beyotime, Beijing, China), gefolgt von Exposition gegenüber den Röntgenaufnahmen. Die Dichte der Proteinbande wurde mit einem Gel-Bildsystem (Tanon, Shanghai, China) quantifiziert und normalisiert ß-Aktin Ebenen, die als Lade Kontrollen verwendet wurde. Limma ( lineare Modelle für die Microarray-Daten) basierte Analyse wurde durchgeführt, um die differentiell exprimierte miRNAs mit einem Cut-off-Wert von mindestens 2-fach Änderungen (FC) mit p <zu identifizieren; 0,05 und FDR < 0,05. SPSS 21.0 Software (SPSS, Chicago, IL, USA) wurde verwendet, Receiver Operating Characteristic (ROC) Kurve und logistische Regressionsanalyse durchzuführen. Die Sensitivität, Spezifität und die Fläche unter der Kurve (AUC) wurden berechnet, um die Med-Calc statistische Software und p-Wert <verwendet wird; 0,05 wurde als statistisch signifikant angesehen. Western-Blot-Daten wurden mit GraphPad Prism Version 5.0 (San Diego, CA, USA) und die Differenz zwischen Tumor und Normalgewebe wurden analysiert nach dem one-tailed Student-t-Test und ein p-Wert <ausgewertet; 0,05 wurde als statistisch signifikant.
. PLoS ONE 10 (4): e0122882. doi: 10.1371 /journal.pone.0122882
Infektion oder den Verbrauch von Räucherwaren) und genetische Faktoren ( E-Cadherin
Mutation), um die Anfälligkeit für Magenkrebs erhöht durch Änderungen in Onkogenen /Tumor-Suppressor-Gene induziert und /oder epigenetische Profil [4]. Veränderung in dieser kritischen Faktoren führt zu abnormal Regulation von Zellwachstum, Apoptose, Differenzierung und somit Karzinogenese fördern. Mehrere Gen regulatorische Netzwerke koordiniert die Umwandlung der normalen Zelle zu einer Tumorzelle und Tumorprogression fahren. Doch bis heute ist das detaillierte Verständnis der zugrunde liegenden mehrere Gen regulatorische Netzwerke in der Pathogenese von Magenkrebs noch definiert werden. der detaillierten molekularen Mechanismus Netzwerk Bestimmung mit Magenkrebs Entwicklung und Progression verbunden sind, könnten das Verständnis der Karzinogenese im Magengewebe zu verbessern, damit Weg für neue und wirksame Strategien zur Prävention, Diagnose und Behandlung von Magenkrebs zu ebnen.
Materialien und Methoden
Profilieren von differentiell exprimierten mRNA und microRNA bei Magenkrebs Gewebe
Bau von TF-Gen, miRNA-Targeting-Gen und TF-miRNA Co-regulatorischer Netzwerke
Funktionsannotationen ausgewählter Gene
Quantitative RT-PCR (qRT-PCR)
Die statistische Analyse
Ergebnisse
 Länder mit älterer Bevölkerung haben höhere SARS-CoV-2-Infektionen und -Todesfälle,
Länder mit älterer Bevölkerung haben höhere SARS-CoV-2-Infektionen und -Todesfälle,
 Große Studie zeigt, dass die Viruslast von SARS-CoV-2 bei Kindern am niedrigsten ist
Große Studie zeigt, dass die Viruslast von SARS-CoV-2 bei Kindern am niedrigsten ist
 Forschung verbindet die Prävalenz von SARS-CoV-2,
Forschung verbindet die Prävalenz von SARS-CoV-2,
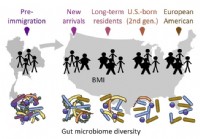 Die Migration beeinflusst die Darmmikrobiota, was sich wiederum auf die Gesundheit auswirkt, finden Forscher
Die Migration beeinflusst die Darmmikrobiota, was sich wiederum auf die Gesundheit auswirkt, finden Forscher
 Forscher setzen Phagentherapie ein, um alkoholische Lebererkrankungen erfolgreich zu behandeln
Forscher setzen Phagentherapie ein, um alkoholische Lebererkrankungen erfolgreich zu behandeln
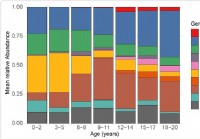 Veränderung des Mikrobioms der oberen Atemwege bei Kindern im Zusammenhang mit der Anfälligkeit für SARS-CoV-2
Veränderung des Mikrobioms der oberen Atemwege bei Kindern im Zusammenhang mit der Anfälligkeit für SARS-CoV-2
 Colitis ulcerosa und eine fehlende Mikrobe im Darm
Colitis ulcerosa ist eine stark schwächende entzündliche Erkrankung des Darms, die zu lähmenden Symptomen führt, die die Lebensqualität stark beeinträchtigen können. Forscher der Stanford University S
Colitis ulcerosa und eine fehlende Mikrobe im Darm
Colitis ulcerosa ist eine stark schwächende entzündliche Erkrankung des Darms, die zu lähmenden Symptomen führt, die die Lebensqualität stark beeinträchtigen können. Forscher der Stanford University S
 Neue Strategie kann die Darm-Hirn-Kommunikation stärken
Das Kommunikationssystem zwischen Darm und Gehirn wird als Darm-Hirn-Achse bezeichnet und ist gut etabliert. Jetzt, Wissenschaftler haben eine Strategie entwickelt, die das Volumen der Darm-Körper-Kom
Neue Strategie kann die Darm-Hirn-Kommunikation stärken
Das Kommunikationssystem zwischen Darm und Gehirn wird als Darm-Hirn-Achse bezeichnet und ist gut etabliert. Jetzt, Wissenschaftler haben eine Strategie entwickelt, die das Volumen der Darm-Körper-Kom
 Kunststoffe, die heute häufig im menschlichen Stuhl zu finden sind
Fast acht Milliarden Tonnen Plastik gelangen jedes Jahr in die Ozeane. Diese riesige Menge Plastik wird entweder an Land gespült oder zerfällt in winzige Stücke mit einem Durchmesser von weniger als 5
Kunststoffe, die heute häufig im menschlichen Stuhl zu finden sind
Fast acht Milliarden Tonnen Plastik gelangen jedes Jahr in die Ozeane. Diese riesige Menge Plastik wird entweder an Land gespült oder zerfällt in winzige Stücke mit einem Durchmesser von weniger als 5