Résumé
carcinome gastrique est l'une des principales causes de la mortalité liée au cancer dans le monde entier. la détection précoce et le traitement conduit à un excellent pronostic chez les patients atteints de cancer gastrique précoce (EGC), alors que le pronostic des patients atteints de cancer gastrique avancé (AGC) reste faible. Il est difficile de savoir si et CGS CAG sont des entités distinctes ou si CGS sont les premiers stades de la CAG. Nous avons effectué le séquençage de l'exome ensemble de quatre échantillons de patients atteints de EGC et comparé les résultats avec ceux de la CAG. Dans les deux CGS et CAG, un total de 268 gènes ont été fréquemment muté et mutations indépendantes ont été en outre trouvé dans CGS (516 gènes) et CAG (3104 gènes). Une fréquence plus élevée de C > G transitions ont été observées dans le type intestinal comparé à diffuser de type carcinomes ( P Citation:. Kang G , Hwang WC, Do IG, Wang K, Kang SY, Lee J, et al. (2013) Exome séquençage Identifie précoce gastrique Carcinome comme un stade précoce de cancer avancé de l'estomac. PLoS ONE 8 (12): e82770. doi: 10.1371 /journal.pone.0082770 Editeur: Patrick Tan, Duke-National University de Singapour Graduate Medical School de Singapour Reçu le 17 Juillet 2013; Accepté le 27 Octobre 2013; Publié le 23 Décembre, 2013 | Droit d'auteur: © 2013 Kang et al. Ceci est un article en accès libre distribué sous les termes de la licence Creative Commons Attribution, qui permet une utilisation sans restriction, la distribution et la reproduction sur tout support, à condition que l'auteur et la source originelle sont crédités Financement:. Cette étude a été soutenue par une subvention de la national Research Foundation de Corée (2012-P4KR 003) et une subvention Institut de recherche biomédicale Samsung (# SBRI-SP1B20111). Les bailleurs de fonds ont joué aucun rôle dans la conception de l'étude, la collecte et l'analyse des données, la décision de publier, ou de la préparation du manuscrit Intérêts concurrents:. KW est employé par Pfizer Inc. Toutefois, cela ne modifie pas l'adhésion de l'auteur tous les PLOS ONE politiques sur les données et les matériaux de partage. Les auteurs ont déclaré qu'il n'y a pas d'autres intérêts divergents existent. carcinome gastrique Introduction (GC) est une maladie hétérogène avec plusieurs étiologies environnementaux, voies alternatives de la cancérogenèse et pas de haute fréquence connue perturbation oncogénique [1], [2], [3]. La classification Lauren est avéré utile dans l'évaluation de l'histoire naturelle de GC, en particulier en ce qui concerne les tendances de l'incidence, les corrélations clinicopathologiques et précurseurs étiologiques [4]. Lauren classifiée adénocarcinome gastrique dans l'intestin et diffus en fonction des caractéristiques morphologiques de la tumeur [4], [5], [6]. carcinomes de type Intestinal sont censés résulter secondaire à une gastrite atrophique chronique associée à H. pylori GC est l'une des principales causes de cancer- la mortalité liée dans le monde entier. détection et de traitement des résultats précoces dans un excellent pronostic pour les patients atteints de cancer gastrique précoce (EGC), alors que le pronostic des patients atteints de cancer gastrique avancé (AGC) reste faible. Cependant, il est difficile de savoir si et CGS CAG sont des entités distinctes ou sont la même tumeur progresse du début à des stades avancés [10]. Les signatures moléculaires qui distinguent EGC d'AGC sont importants pour faciliter l'identification de nouveaux marqueurs pronostiques et cibles thérapeutiques potentielles. Récemment, le séquençage de l'exome dans 22 [11] et 15 [12] échantillons AGC a montré des mutations inactivant fréquentes dans l'adhésion cellulaire et remodelage de la chromatine des gènes, et les altérations génétiques diffèrent entre les sous-groupes stratifiés par le virus d'Epstein-Barr (EBV) ou H. pylori Préparation d'échantillons Matériaux et méthodes tumeur et tissus gastriques non néoplasiques ont été recueillies à partir d'échantillons de gastrectomie. La présente étude a été réalisée après l'approbation du conseil d'Samsung Medical Center Institutional Review, et tous les patients ont signé un consentement éclairé avant la chirurgie. Pour les échantillons de tumeurs, masses étaient > 4 cm sur l'inspection brute, et la muqueuse de surface de chaque tumeur a été obtenue. Après l'incorporation dans l'OCT médias, le tissu a été coupé et H & E tachée. Des échantillons de > le contenu de la tumeur de 90% ont été sélectionnés pour l'extraction d'ADN avec un kit Mini (Qiagen, Valencia, CA, USA) et traité avec de la RNase A pour évacuer le reste de l'ARN. L'ADN a été également extrait du tissu gastrique non affecté apparié, qui a été obtenu à partir du site éloigné de la tumeur et confirmé comme étant exemptes de tumeur. MSI a été analysé avec cinq marqueurs NCI comme décrit précédemment [13]. La présence de l'EBV a été détectée par l'ARN d'EBV codées L'hybridation in situ de la manière décrite précédemment, et seuls les cas le signal fort au sein de la quasi-totalité des noyaux de cellules tumorales ont été considérés comme positifs [14]. Des détails supplémentaires pour les échantillons EGC sont fournis dans le tableau 1. enrichissement d'Exome et le séquençage Exome enrichissement (Kit SureSelect Tous Human Exon, Agilent Technologies) et les bibliothèques de séquençage Illumina ont été préparées selon le fabricant instructions. En bref, 3 pg d'ADN génomique a été cisaillé avec le système Covaris S2; les fragments d'ADN ont été réparées bout, prolongé par un «A» sur la base de l'extrémité 3 ', ligaturé avec des adaptateurs de fin paires et amplifié (quatre cycles). bibliothèques adaptateur ligaturé contenant Exome-ont été hybrides pendant 24 h avec biotinylés appâts oligo-ARN et enrichi avec des billes magnétiques streptavidine conjugué. Les bibliothèques finales ont été amplifiés par 11 cycles de PCR, et soumis à un séquençage Illumina sur une piste du séquenceur HiSeq 2000 avec un insert de taille ciblée de ~180 pb. Tout le séquençage a été exécuté avec jumelé fin 65 pb lit et a été effectuée selon le protocole standard de Ilumina. En moyenne, la pureté ~136.3 millions filtré lectures ont été générées pour chaque échantillon. Le pourcentage moyen de double lit en raison de PCR et d'artefacts optiques était de 0% dans notre ensemble de données, et ~123.7 millions mappé uniquement les lectures ont été obtenues pour chaque échantillon. En moyenne, 69,1% de lectures dans chaque échantillon a au moins 50% de recouvrement avec une région cible de ± 100 pb dans la bibliothèque exome d'appât SureSelect entier. Les régions ciblées dans chaque échantillon ont été séquencés à une profondeur moyenne de 113,7 ×, avec ~98.8% des régions ciblées couvertes ≥1 ×, ~94.3% ≥10 ×, ~82.4% ≥30 ×, ~70.8% ≥50 ×, ~66.4% ≥60 ×, ~62.2% ≥70 ×, ~58.2% ≥80 ×, ~54.4% ≥90 × et ~50.8% ≥100 x. Des résumés détaillés de la qualité des données brutes sont décrites dans le tableau S1. A titre de comparaison, le même algorithme (SMART), utilisé dans l'ensemble de données précédent d'échantillons AGC [11], a été appliquée à ces données pour identifier somatiques des variations et des insertions /délétions (indels) des altérations de données de séquence courte de lecture d'un seul nucléotide. L'ensemble de données a été déposé dans les archives européennes Nucleotide et peut être consulté à http://www.ebi.ac.uk/ena/data/view/PRJEB 4850. Mutations détectées par le séquençage de l'exome étaient encore validée par un séquençage Sanger et PCR. En bref, des amorces sont conçues en utilisant le logiciel Primer3 (http://frodo.wi.mit.edu) et les séquences sont listées dans le tableau S3. Les produits amplifiés par PCR ont ensuite été séquences en utilisant un kit BigDye Terminator Cycle v3.1 de séquençage et un séquenceur automatisé ABI 3700 (Applied Biosystems, Foster City, CA, USA). Résultats altérations CGS au total, 2.389 mutations somatiques ont été identifiées dans les quatre échantillons EGC, dont 1117 se sont produits dans des régions ou des sites d'épissage essentiels (627 faux-sens, 32 non-sens, 10 site d'épissage essentiel, 169 indels et 279 de codage synonyme) (figure 1 et les tableaux 2 et S2). Un GC avec MSI haute avait 727 mutations non silencieuses, y compris les gènes de réparation des mésappariements ( MSH6 Comparaison entre EGC et AGC Pour la comparaison de nos résultats sur EGC avec ceux de CAG, deux données de séquençage de l'exome ensemble récemment publiées ont été utilisées [11], [12]. Wang et al dans 37 AGC et 4 échantillons EGC, mutations non silencieuses (faux-sens, non-sens, et le site d'épissage indels essentiel) ont été détectés dans 3.372 et 784 gènes, respectivement. Dans les deux CGS et CAG, 268 gènes ont été fréquemment mutés; BCORL1, LRP2, LRP12, macf1, PRKCI Discussion Bien que le séquençage de l'exome ensemble a été rapporté pour 37 échantillons AGC [11], [12], il n'y a pas eu une telle étude pour évaluer la carcinogenèse précoce au niveau génétique. Pour explorer le répertoire complet des mutations somatiques dans CGS, nous avons réalisé le séquençage de l'exome ensemble de quatre échantillons EGC appariés, et nous avons trouvé des signatures génétiques distinctes et communes entre CGS et CAG qui peuvent identifier les gènes impliqués dans la carcinogenèse précoce et la progression ultérieure. cancers épithéliaux ont souvent variables spectres de mutation pointant vers particulier stimuli mutagènes [15], [16]. Par exemple, des taux élevés de A > C et C > T transitions ont été observées dans les adénocarcinomes de l'œsophage et les mélanomes exposées au soleil, respectivement, ce qui suggère que ces mutations sont attribuables au reflux gastro-oesophagien et l'exposition aux ultraviolets [15], [17]. Une étude de séquençage du génome entier précédent dans deux adénocarcinomes gastriques montré fréquents C > A et T > A modifications par rapport aux génomes normaux [18]. Ici, nous avons trouvé fréquents C > G transitions dans les carcinomes de type intestinal par rapport à diffuser de type GCS après exclusion de MSI-haute GCS. Nos bons de souscription d'observation uniques futures études visant à définir l'étiologie spécifique qui contribue potentiellement à la compréhension des voies moléculaires complexes et mal connus de glucocorticoïdes de type intestinal. Grâce à une analyse comparative, nous avons identifié 268 gènes qui se chevauchent avec des mutations non silencieuses partagées par les deux axes libres et CAG (figure 3). Environ un tiers des mutations non silencieuses en CGS sont partagés avec CAG et 8% des mutations non silencieuses trouvées dans CAG sont partagés avec CGS. Une étude antérieure et une analyse de l'expression génique a montré que la majorité des altérations associées aux axes libres sont retenus dans CAG et d'autres modifications d'expression marquent le passage de l'EGC AGC [10]. Dans l'ensemble, ces résultats indiquent que EGC représente un stade moléculaire précoce des AGC, et les gènes mutés couramment jouent un rôle important dans la progression de EGC à AGC. Nous avons réaffirmé que TP53 Bien que la prévalence des mutations récurrentes dans CGS était relativement faible, 13 gènes ont été mutés dans au moins deux échantillons, et a eu très peu synonymes, mutations introniques et /ou non traduites. Parmi ces 13 gènes, DYRK3, GPR116, MCM10, PCDH17, PCDHB1, RDH5 La perte de fonction dans les molécules d'adhésion cellulaire augmente la capacité des cellules tumorales à envahir les tissus environnants, et un dysfonctionnement dans un complexe de remodelage de la chromatine favorise l'instabilité chromosomique qui entraîne la tumorigenèse [27]. Aucun de nos échantillons EGC avait des mutations de protéines modifiant des gènes de remodelage de la chromatine trouvés dans CAG, tels que ARID1A, MLL3, PBRM1 dans l'ensemble, notre étude suggère que EGC et AGC partagent des mutations somatiques communs, et AGC est associée à des altérations génétiques cumulatifs supplémentaires dans l'adhésion cellulaire et des gènes de remodelage de la chromatine. Les signatures moléculaires qui distinguent EGC d'AGC sont importantes pour aider à identifier de nouveaux marqueurs pronostiques et cibles thérapeutiques potentielles. Des études plus importantes sont nécessaires pour déterminer l'importance biologique des gènes mutés dans récurrente CGS.
= 0,010). Le DYRK3, GPR116, MCM10, PCDH17, PCDHB1, RDH5
et Les gènes UNC5C de sont mutés dans récurrente et CGS peuvent être impliqués dans la carcinogenèse précoce
et métaplasie intestinale [7]. Diffuse-Type glucocorticoïdes ne sont pas associés à la métaplasie intestinale et peuvent provenir de mutations unicellulaires dans les glandes gastriques normales [4], [8], [9].
infection et l'instabilité des microsatellites (MSI) statut. Pour explorer davantage les altérations génétiques sous-jacentes GCS, nous avons réalisé le séquençage de l'exome entier dans quatre paires de EGC et le tissu normal, et comparé les résultats à ceux de la CAG.
somatique
et MSH3
), alors que les trois stables microsatellite (MSS) échantillons avaient une moyenne de 37 ans, différence d'environ 20 fois. Les rapports non synonymes à synonymes dans les cancers du SMS avaient tendance à être plus élevé que celui de la MSI haute cancer, mais la différence était statistiquement non significatif. C > T et G > A transitions étaient la mutation la plus fréquente (61%) dans le CGS, et il n'y avait pas de différence significative dans les changements de paires de bases simples entre MSI-haut et les cancers du SMS (Figure 2A et Tableau S4). 784 gènes présentant des mutations non silencieuses, 13 ont été mutés en deux ou plusieurs échantillons. Ces gènes inclus connus pour être impliqués dans la carcinogenèse gastrique ( TP53
) et rapporté dans le catalogue de Somatic Mutations dans le Cancer (COSMIC) à muter en glucocorticoïdes ( DYRK3, MCM10, PCDH17
et UNC5C
) (tableau 3). Parmi les gènes sélectionnés pour la validation, PCDH17 de la mutation a été très probablement pas validé par la méthode de Sanger en raison des basses fréquences de l'allèle mutant (tableau S3). Fait intéressant, dans un EGC de type diffus avec MSI-haute, un EGFR
(c.2224G > A, p.V742I). Mutation a été identifiée
. détectée 164 non-silencieux et 48 mutations synonymes en moyenne dans 22 échantillons AGC avec 116 × profondeur de couverture moyenne [11]. Zang et al
. détecté en moyenne 50 non-silencieuse et 16 mutations somatiques synonymes dans 15 échantillons AGC avec 96 × profondeur de couverture moyenne [12]. En comparaison directe entre les quatre axes libres et 37 CAG, il n'y avait pas de différence significative dans le nombre de type de mutation (Figure 1). Les changements de paires de bases simples dans CGS étaient semblables à un rapport précédent par Wang et al
. [11], montrant un nombre nettement plus élevé de C > T et G > A transitions dans les deux MSS et les tumeurs MSI-haut (figure 2A et le tableau S4). Fait intéressant, C > G transitions étaient plus fréquents dans de type intestinal que dans GCS-type diffuse dans tous les échantillons du SMS, qui comprenait trois axes libres et 18 CAG (test de Wilcoxon rang somme, P
= 0,010) (figure 2B et le tableau S4).
et Les gènes TP53 de ont été mutés dans au moins deux échantillons EGC et le ACVR2A, CCNL1, CTNNB1, FMN2, PTEN, RPL22
et TTN
gènes, ainsi que d'autres, étaient significativement associés à CAG avec un taux de fausse découverte de < 0,2 [11], [12] (figure 3). Analyse d'annotation fonctionnelle utilisant DAVID (http://david.abcc.ncifcrf.gov) pour examiner les gènes trouvés chevauchement entre les deux ensembles d'échantillons a révélé que les termes significativement enrichi inclus actine, cytosquelette, la projection de la cellule et de la jonction cellule-cellule (tableau S5).
est le gène le plus fréquemment muté dans GCS, avec TP53 de les mutations trouvées dans la moitié des EGC et les deux tiers des échantillons AGC. Parmi les gènes qui se chevauchent, AKAP9, CAMTA1, COL1A1, CTNNB1, KDM5A
et RPL22
ont été annotées comme oncogènes, alors que ATM, FBXW7, MSH6, NF1, PTEN, SETD2
et TP53
étaient des gènes suppresseurs de tumeurs par le gène de recensement Sanger (http://cancer.sanger.ac.uk/cancergenome/projects/census). Parmi les gènes du cancer du recensement, nous avons d'abord identifié une EGFR
mutation (c.2224G > A, p.V742I) dans un diffus de type EGC avec MSI-haute. Dans une étude récente sur 63 MSI haute GCS, EGFR
mutation n'a pas été détectée par séquençage direct du domaine kinase (exons 18, 19, 20 et 21) [19]. La même mutation V742I a été rapportée chez un patient souffrant d'un cancer de l'endomètre et dans une lignée cellulaire de gliome [20], [21]. La signification clinique de cette mutation rare doit être validée dans un proche avenir.
et UNC5C peut être spécifique pour GC à un stade précoce, ce qui suggère un rôle possible dans le début des années cancérogenèse. Dans notre série, Les mutations PCDH17 se sont produits dans des glucocorticoïdes de type intestinal avec MSS, y compris un échantillon de EBV-positive. Précédent analyses génomiques globales de cancer colorectal et du pancréas ont également révélé des mutations faux-sens dans certains membres de PCDH
(protocadherin) subfamilias [22], [23]. Cependant, les mutations détectées dans nos CGS par séquençage Illumina n'a pas été confirmé par séquençage Sanger, probablement parce que les fréquences des allèles mutants étaient très faibles. UNC5C appartient à la famille des récepteurs de dépendance fonctionnelle, dont les membres partagent la capacité d'induire l'apoptose en l'absence de leurs ligands [24], [25]. méthylation aberrante de ce gène a été rapporté au cours de la carcinogenèse gastrique, ce qui a disparu dans la méthylation GCS très avancés [26]. Pour les gènes restants, leur pertinence fonctionnelle dans GC reste incertaine.
et MBD2
[11], [12], ce qui suggère la chromatine modification intervient tard dans la progression de la GC.
Informations complémentaires
Tableau S1.
Résumé des statistiques de séquençage de l'exome entier pour quatre paires d'échantillons gastriques
doi:. 10.1371 /journal.pone.0082770.s001
(PDF)
Tableau S2.
Liste de toutes les mutations somatiques dans les cancers gastriques précoces identifiés par séquençage de l'exome
doi:. 10.1371 /journal.pone.0082770.s002
(XLS)
Tableau S3.
Amorces pour le séquençage Sanger, la fréquence des allèles et des résultats de validation
doi: 10.1371. /journal.pone.0082770.s003
(PDF)
Tableau S4. Comparaison des spectres point de mutation somatique dans les cancers gastriques par l'état de la tumeur et le type histologique
doi:. 10.1371 /journal.pone.0082770.s004
(PDF)
Tableau S5.
DAVID analyse des voies des 268 gènes qui se chevauchent entre les cancers précoces et avancés gastriques
doi:. 10.1371 /journal.pone.0082770.s005
(XLS)
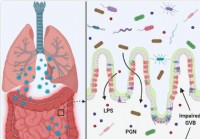 Les fuites intestinales et la dysbiose microbienne pourraient contribuer à la tempête de cytokines dans les cas de COVID-19 gravement malades
Les fuites intestinales et la dysbiose microbienne pourraient contribuer à la tempête de cytokines dans les cas de COVID-19 gravement malades
 Les probiotiques comme thérapie adjuvante pour les patients COVID-19
Les probiotiques comme thérapie adjuvante pour les patients COVID-19
 Ce que vous mangez peut changer la façon dont les antibiotiques affectent votre intestin
Ce que vous mangez peut changer la façon dont les antibiotiques affectent votre intestin
 Faits saillants et principaux points à retenir du Boston Bacterial Meeting (BBM) 2019
Faits saillants et principaux points à retenir du Boston Bacterial Meeting (BBM) 2019
 Mesures pour prévenir la transmission du SRAS-CoV-2 par les eaux usées dans les régions pauvres
Mesures pour prévenir la transmission du SRAS-CoV-2 par les eaux usées dans les régions pauvres
 La qualité du sommeil pourrait être un indicateur des découvertes ultérieures de la maladie d'Alzheimer
La qualité du sommeil pourrait être un indicateur des découvertes ultérieures de la maladie d'Alzheimer
 Une nouvelle étude pourrait aider à prévenir les infections mortelles chez les bébés
Les nourrissons prématurés nés avant 28-30 semaines de vie sont à haut risque de nombreuses complications, parmi lesquelles les chances de mourir dune infection débutant dans lintestin sont très grand
Une nouvelle étude pourrait aider à prévenir les infections mortelles chez les bébés
Les nourrissons prématurés nés avant 28-30 semaines de vie sont à haut risque de nombreuses complications, parmi lesquelles les chances de mourir dune infection débutant dans lintestin sont très grand
 La découverte de 100 nouveaux gènes pourrait aider la recherche sur les maladies de la pigmentation
Une nouvelle étude menée par des chercheurs du Kings College de Londres et du centre médical universitaire Erasmus MC de Rotterdam a découvert 124 gènes qui jouent un rôle clé dans la détermination de
La découverte de 100 nouveaux gènes pourrait aider la recherche sur les maladies de la pigmentation
Une nouvelle étude menée par des chercheurs du Kings College de Londres et du centre médical universitaire Erasmus MC de Rotterdam a découvert 124 gènes qui jouent un rôle clé dans la détermination de
 Les microbiomes anciens des primates pourraient fournir plus d'informations sur le développement humain
Les microbiomes humains anciens sont sous le microscope pour ce quils disent aux scientifiques sur les gens dil y a longtemps. Une nouvelle étude publiée dans la revue Frontières en écologie et évolu
Les microbiomes anciens des primates pourraient fournir plus d'informations sur le développement humain
Les microbiomes humains anciens sont sous le microscope pour ce quils disent aux scientifiques sur les gens dil y a longtemps. Une nouvelle étude publiée dans la revue Frontières en écologie et évolu