Magenkarzinom ist eine der Hauptursachen für Krebs-Mortalität weltweit. Früherkennung und Behandlung führt zu einer exzellenten Prognose bei Patienten mit Magenfrühkarzinomen (EGC), während die Prognose von Patienten mit fortgeschrittenem Magenkrebs (AGC) schlecht bleibt. Es ist unklar, ob EGCs und AGCs sind getrennte Einheiten oder ob EGCs sind die Anfangsphasen der AGCS. Wir führten ganze Exoms Sequenzierung von vier Proben von Patienten mit EGC und verglichen die Ergebnisse mit denen aus AGCs. In beiden EGCs und AGCS wurden insgesamt 268 Gene häufig mutiert und unabhängige Mutationen wurden zusätzlich in EGCs (516 Gene) und AGCs (3104 Gene) gefunden. Eine höhere Frequenz von C > G Übergänge wurde in Darm-Typ beobachtet im Vergleich zu diffundieren artigen Karzinomen ( P Citation:. Kang G Hwang WC, Do IG, Wang K, Kang SY, Lee J, et al. (2013) Exome Sequencing Identifiziert frühen Magenkarzinom als einem frühen Stadium des fortgeschrittenen Magenkarzinoms. PLoS ONE 8 (12): e82770. doi: 10.1371 /journal.pone.0082770 Editor: Patrick Tan, Duke-National University of Singapore Graduate Medical School, Singapur Empfangen: 17. Juli 2013; Akzeptiert: 27. Oktober 2013 beginnen; Veröffentlicht: 23. Dezember 2013 Copyright: © 2013 Kang et al. Dies ist eine Open-Access-Artikel unter den Bedingungen der Lizenz Creative Commons, die uneingeschränkte Nutzung erlaubt, die Verteilung und Vervielfältigung in jedem Medium, vorausgesetzt, der ursprüngliche Autor und Quelle genannt werden Finanzierung:. Diese Studie wurde durch einen Zuschuss von der National Research Foundation of Korea (2012-P4KR 003) und einem Samsung Biomedical Research Institute Zuschuss (# SBRI-SP1B20111) unterstützt. Die Geldgeber hatten keine Rolle in Studiendesign, Datenerfassung und Analyse, Entscheidung oder Vorbereitung des Manuskripts zur Veröffentlichung konkurrierende Interessen. KW wird jedoch von Pfizer Inc. beschäftigt, ist dies nicht des Autors ändert die Einhaltung alle PLoS ONE Politik auf den Austausch von Daten und Materialien. Die Autoren haben erklärt, dass keine anderen konkurrierenden Interessen bestehen. Magenkarzinom (GC) ist eine heterogene Erkrankung mit mehreren Umweltkrankheitsursachen, alternative Wege der Karzinogenese und ohne bekannte Hochfrequenz-onkogenen Störung [1], [2], [3]. Die Lauren Klassifikation hat sich als nützlich erwiesen, um die Naturgeschichte der GC bei der Bewertung, vor allem im Hinblick auf die Inzidenz-Trends, klinisch-pathologische Korrelationen und ursächliche Vorstufen [4]. Lauren klassifiziert Adenokarzinom des Magens in Darm- und diffundieren nach morphologischen Eigenschaften des Tumors [4], [5], [6]. Darm-Typ-Karzinome sind vermutlich sekundär entstehen chronische atrophische Gastritis im Zusammenhang mit H. pylori GC ist eine der wichtigsten Ursachen von Krebs- im Zusammenhang mit weltweiten Sterblichkeit. Früherkennung und Behandlung führt zu einer exzellenten Prognose für Patienten mit Magenfrühkarzinomen (EGC), während die Prognose von Patienten mit fortgeschrittenem Magenkrebs (AGC) schlecht bleibt. Es ist jedoch unklar, ob EGCs und AGCs sind getrennte Einheiten oder sind die gleichen Tumor von früh bis fortgeschrittenen Stadien voran [10]. Die molekularen Signaturen EGC von AGC zu unterscheiden sind wichtige Identifizierung neuer prognostischer Marker und potentielle therapeutische Targets zu unterstützen. Vor kurzem Exoms Sequenzierung in 22 [11] und 15 [12] AGC Proben zeigten häufige inaktivierende Mutationen in der Zelladhäsion und Chromatin-Remodeling-Gene und die genetischen Veränderungen bei den Untergruppen geschichtet durch das Epstein-Barr-Virus (EBV) oder H unterschieden. pylori Probenvorbereitung Tumor und nicht-neoplastische Magengewebe wurden von Gastrektomie Proben gesammelt. Die vorliegende Studie wurde nach der Genehmigung vom Institutional Review Board von Samsung Medical Center durchgeführt, und alle Patienten gaben Zustimmung vor der Operation informiert geschrieben. Für Tumorproben, Massen waren > 4 cm auf grobe Inspektion und die Oberfläche Schleimhaut von jedem Tumor beschafft wurde. Nachdem in OCT Medien Einbettung wurde das Gewebe geschnitten und H & E gefärbt. Proben von > 90% tumorGehalt zur DNA-Extraktion mit einem Mini Kit (Qiagen, Valencia, CA, USA) und mit RNase A wurden ausgewählt verbleibenden RNA zu entfernen. DNA wurde auch aus paarigen unbeeinflußt Magengewebe extrahiert, das von der Tumorstelle entfernten erhalten wurde und bestätigt tumorfrei. MSI wurde mit fünf NCI Marker analysiert, wie zuvor beschrieben [13]. Die Anwesenheit von EBV wurde durch EBV-kodierte RNA in situ Exome Anreicherung und Sequenzierung Exome Anreicherung (Sureselect Menschliches, Exon Kit, Agilent Technologies) und Illumina-Sequenzierung Bibliotheken wurden nach dem Hersteller vorbereitet Anleitung. Kurz gesagt, wurden 3 ug genomische DNA mit dem Covaris S2 System geschert; die DNA-Fragmente wurden Ende repariert, mit einem "A" Basis am 3'-Ende verlängert, mit Paired-End-Adapter ligiert und verstärkt (vier Zyklen). Exome haltige Adapter-ligierten Bibliotheken wurden mit biotinyliertem Oligo-RNA Köder für 24 h hybridisiert und mit Streptavidin-konjugierten magnetischen Kügelchen angereichert. Die endgültigen Bibliotheken wurden weiter verstärkt durch 11 PCR-Zyklen und auf einer Spur der HiSeq 2000 Sequenzer mit einer gezielten Insertgröße von ~ 180 bp bis Illumina-Sequenzierung unterzogen. Alle Sequenzierung wurde durchgeführt, mit Paired-End 65-bp liest und wurde nach Ilumina Standardprotokoll durchgeführt. Im Durchschnitt ~136.3 Millionen Reinheit filterten liest wurden für jede Probe erzeugt. Der mittlere Prozentsatz der doppelten liest aufgrund PCR und optische Artefakte von 0% in unserem Datensatz und ~123.7 Millionen eindeutig abgebildet liest für jede Probe erhalten wurden. Im Durchschnitt liest 69,1% von mindestens 50% Überlappung hatte mit jedem Zielbereich ± 100 bp in der Sureselect ganze Exoms bait Bibliothek in jeder Probe. Die Zielregionen in jeder Probe zu einer durchschnittlichen Tiefe von 113,7 ×, bedeckt mit ~98.8% der Zielregionen wurden sequenziert ≥1 ×, ~94.3% ≥10 ×, ~82.4% ≥30 ×, ~70.8% ≥50 ×, ~66.4% ≥60 ×, ~62.2% ≥70 ×, ~58.2% ≥80 ×, ~54.4% ≥90 × und ~50.8% ≥100 ×. Ausführliche Zusammenfassungen der Rohdaten Qualität sind in Tabelle S1 beschrieben. Zum Vergleich wurde der gleiche Algorithmus (SMART), in dem vorherigen Datensatz von AGC Proben verwendet [11] wurde auf diese Daten angewendet, um somatische single-nucleotide Variationen und Insertionen /Deletionen (indels) Änderungen von kurzen gelesenen Sequenzdaten identifizieren. Der Datensatz hat in der European Nucleotide Archive hinterlegt und kann bei http://www.ebi.ac.uk/ena/data/view/PRJEB 4850. Mutationen durch Exoms Sequenzierung nachgewiesen wurden weiter durch PCR und Sanger-Sequenzierung bestätigt. Verwendung Primer3 Software (http://frodo.wi.mit.edu), und die Sequenzen sind in Tabelle S3 Kurz gesagt, werden Primer entworfen. Die PCR-amplifizierte Produkte wurden dann ein Kit v3.1 Cycle Sequencing BigDye Terminator sequenziert und mit einem ABI 3700 automatisierten Sequenzer (Applied Biosystems, Foster City, CA, USA).
= 0,010). Die DYRK3, GPR116, MCM10, PCDH17, PCDHB1, RDH5
und UNC5C
Gene sind wiederkehrend in EGCs mutiert und kann in der frühen Karzinogenese beteiligt sein
Einführung
und intestinale Metaplasie [7]. Diffuse-Typ GCs sind nicht mit intestinaler Metaplasie verbunden und kann von Einzelzellen-Mutationen innerhalb der normalen Magendrüsen entstehen [4], [8], [9].
Infektion und Mikrosatelliteninstabilität (MSI) Status. Um die genetischen Veränderungen erkunden GCs zugrunde liegen, führten wir ganze Exoms Sequenzierung in vier paarweise von EGC und normalem Gewebe, und verglichen die Ergebnisse mit denen aus AGCs.
Materialien und Methoden
Hybridisierung nachgewiesen, wie vorher beschrieben, und nur die Fälle mit starkem Signal innerhalb fast alle der Tumorzellkerne wurden positive [14] betrachtet. Weitere Einzelheiten für die EGC-Proben sind in Tabelle 1
bereitgestellt
zugegriffen werden
Ergebnisse
 Dass Pepto Ihrem Geschwür wahrscheinlich nicht hilft
Dass Pepto Ihrem Geschwür wahrscheinlich nicht hilft
 Spermienmikrobiom mit RNA-Sequenzierung entdeckt
Spermienmikrobiom mit RNA-Sequenzierung entdeckt
 Studie gibt Aufschluss über die Ursachen von schwächenden Darmschmerzen
Studie gibt Aufschluss über die Ursachen von schwächenden Darmschmerzen
 Neue Forschung identifiziert einen Zusammenhang zwischen dem Darmmikrobiom und Schlaganfällen
Neue Forschung identifiziert einen Zusammenhang zwischen dem Darmmikrobiom und Schlaganfällen
 Neue Studie könnte helfen, tödliche Infektionen bei Babys zu verhindern
Neue Studie könnte helfen, tödliche Infektionen bei Babys zu verhindern
 Sind DNA-basierte Diäten und personalisierte medizinische Lebensmittel die Zukunft für die Gewichtsabnahme?
Sind DNA-basierte Diäten und personalisierte medizinische Lebensmittel die Zukunft für die Gewichtsabnahme?
 Nahrung beeinflusst selektiv Darmmikroben, Studienergebnisse
Nahrung könnte eine Rolle bei der Bildung der mikrobiellen Flora im menschlichen Darm spielen. Dies wurde in mehreren Studien und Forschungen immer wieder gezeigt. Jetzt haben Forscher der San Diego S
Nahrung beeinflusst selektiv Darmmikroben, Studienergebnisse
Nahrung könnte eine Rolle bei der Bildung der mikrobiellen Flora im menschlichen Darm spielen. Dies wurde in mehreren Studien und Forschungen immer wieder gezeigt. Jetzt haben Forscher der San Diego S
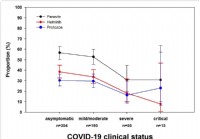 Untersuchungen zeigen, dass ein Befall mit Darmparasiten den Schweregrad von COVID-19 verringert
Wir lernen jeden Tag mehr über die COVID-19-Krankheit. Erwachsene jeden Alters mit bestimmten Grunderkrankungen haben ein erhöhtes Risiko für schwere Erkrankungen durch das Virus, das COVID-19 verursa
Untersuchungen zeigen, dass ein Befall mit Darmparasiten den Schweregrad von COVID-19 verringert
Wir lernen jeden Tag mehr über die COVID-19-Krankheit. Erwachsene jeden Alters mit bestimmten Grunderkrankungen haben ein erhöhtes Risiko für schwere Erkrankungen durch das Virus, das COVID-19 verursa
 Morbus Crohn
Morbus Crohn verursacht eine Entzündung des Magen-Darm-Trakts. Es kann mit Colitis ulcerosa und Reizdarmsyndrom verwechselt werden. aber Morbus Crohn ist einzigartig. An der Ogden Clinic GI at McKay,
Morbus Crohn
Morbus Crohn verursacht eine Entzündung des Magen-Darm-Trakts. Es kann mit Colitis ulcerosa und Reizdarmsyndrom verwechselt werden. aber Morbus Crohn ist einzigartig. An der Ogden Clinic GI at McKay,